|
x |
x |
|
 |
 |
|
INFECTIOUS
DISEASE |
BACTERIOLOGY |
IMMUNOLOGY |
MYCOLOGY |
PARASITOLOGY |
VIROLOGY |
|
PORTUGUESE |
PARASITOLOGY -
CHAPTER THREE
MOLECULAR PARASITOLOGY :
TRYPANOSOMES
EUCARYOTIC CELLS WITH A DIFFERENT WAY OF DOING
THINGS
Dr Richard Hunt
Professor Emeritus
University of South Carolina School of Medicine
|
|
SHQIP - ALBANIAN |
Let us know what you think
FEEDBACK |
|
SEARCH |
|
|

 |
|
Logo image © Jeffrey
Nelson, Rush University, Chicago, Illinois and
The MicrobeLibrary |
|
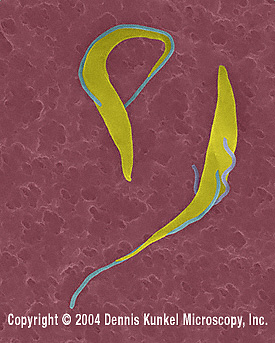
Trypanosome trypomastigotes (Trypanosoma
spp.).
©
Dennis Kunkel Microscopy, Inc.
Used with permission
 |
In this section, we
shall look at the disease of trypanosomiasis and the life cycle of the parasites that
cause it. The novel aspects of the life cycle lead us to ways in which the parasite has
gained unusual biochemical pathways to cope with its niche. These offer potential sites
for chemotherapy.
Trypanosomes are successful parasites which manage to escape the
host's immune response; this happens by a very complex
mechanism of antigen switching and it is the knowledge of this mechanism that has led us
to the first steps in developing an anti-trypanosome vaccine.
TRYPANOSOMES
Trypanosomes belong to the order KINETOPLASTIDA,
so-called because of the large
DNA-containing structure, the kinetoplast, found at the base of the flagellum.
|
| |
|
DISEASES
CAUSED BY TRYPANOSOMES |
|
Trypanosoma
species |
|
Species |
Distribution |
Species exhibiting disease |
Vector |
Disease |
Treatment |
| Trypanosoma brucei gambiense
|
West Africa |
humans |
Glossina palpalis (Tsetse fly)

© OhioState University,
College of Biological Sciences
|
Trypanosomiasis (Sleeping sickness)
chronic
|
 Winterbottoms's sign (CDC)
Winterbottoms's sign (CDC) |
Suramin, pentamidine and berenil: can cure infection if given
before invasion of nervous system. Difluoromethylornithine (DMFO) even if the parasite has invaded the brain. |
|
Trypanosoma brucei rhodesiense |
East Africa |
humans |
Glossina morsitans |
Trypanosomiasis (Sleeping sickness)
acute |
 Patient with advanced sleeping sickness (CDC)
Patient with advanced sleeping sickness (CDC) |
As above |
| Trypanosoma brucei brucei |
Africa |
cattle, antelope, horses, camels |
Glossina pallidipes |
Nagana
acute |
 Milking cow with nagana © WHO
Milking cow with nagana © WHO |
|
| Trypanosoma congolense |
Africa |
cattle, antelope, horses, camels |
Glossina morsitans |
Nagana
chronic |
|
|
| Trypanosoma vivax |
Africa |
cattle, antelope, horses, camels |
Glossina morsitans |
Nagana
acute |
|
|
| Trypanosoma equiperdum |
Africa |
horses, donkeys |
None (transmitted during coitus) |
Dourine
acute |
|
|
| Trypanosoma evansi |
Africa |
camels, horses, deer |
Tabanid fly

From Rob
Hutchinson
|
Sura |
|
|
| Trypanosoma equinum |
|
camels, horses, deer |
Tabanid fly |
Malde Caderas |
|
|
| Trypanosoma cruzi |
South and Central America |
|
Reduvid bugs: Rhodnius prolixus

© OhioState University,
College of Biological Sciences
|
Chagas disease |
|
No effective treatment. Available drugs only kill extracellular parasites.
Most successful treatment
during acute phase - about a 60% success. Benznidazole and Nifurtinox:
current drugs of choice. Required daily for up to 2 months or
more. Hospitalization may be needed because of adverse effects |
|
Leishmania
species |
| Leishmania donovani |
Mediterranean, Africa, India, China, South America |
humans |
Old world: Sandfly (Phlebotomus sp)

(© Tom
Evans)
New world: Lutzomyia chagasi
|
Kal-Azar (Dum-Dum fever) Visceral leishmaniasis |
|
Antimonial compounds |
| Leishmania tropica |
Mediterranean, Africa, India |
humans |
Sandfly (Phlebotomus sp) |
Dermal leishmaniasis (tropical sore oriental sore) |
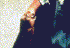
Leishmanial skin lesion © Dr
J. Carl Fox, University of Oklahoma (used with permission)
|
Only effective treatment is antimonial drugs
- severe toxic side effects. Pentostam and Glucantime often used.
Aminosidine effective for cutaneous leishmaniasis and better tolerated. |
| Leishmania major |
Africa, Middle East, India |
humans |
Sandfly (Phlebotomus sp) |
Dermal leishmaniasis (tropical sore) |
|
Antimonial compounds |
| Leishmania braziliensis
L. mexicana
|
South America |
humans |
Lutzomyia sp |
Cutaneous leishmaniasis - skin and mucous membrane lesions,
polyps |

Mouth lesion
(© Tom Evans)
 Nasla mucosa lesion (© Tom
Evans)
Nasla mucosa lesion (© Tom
Evans)
|
Antimonial compounds |
|
| |
|
 Figure 1. Tsetse fly: the vector for African trypansomiasis ©
Ohio State University, College of Biological Sciences
Figure 1. Tsetse fly: the vector for African trypansomiasis ©
Ohio State University, College of Biological Sciences
Figure 2 Winterbottom's sign
(CDC from
Parasites
on the web)
|
AFRICAN TRYPANOSOMIASIS
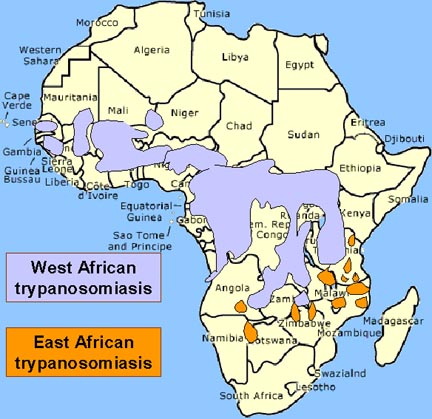
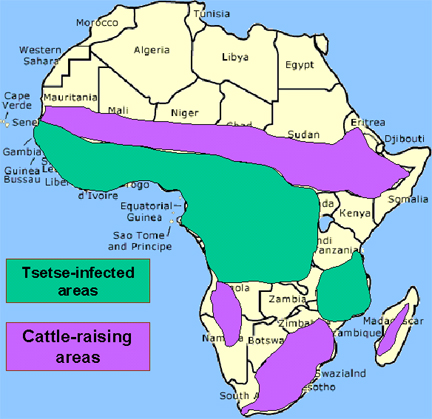
The vector for African trypanosomiasis is
theTsetse fly (figure 1) and the distribution of the disease
parallels the distribution of the vector (figure 4).
The symptoms of African
trypansomiasis depend on host and the sub-species of trypanosome. In T.
gambiense infections there is massive stimulation of immune system and
complement-mediated lysis of host cells (gives characteristic anemia). Generalized pain,
weakness, cramps and swelling of neck lymph nodes (Winterbottom=s sign, figure
2). Parasites invade all organs of the body
including heart and CNS. The latter leads to apathy, mental dullness, tremors, convulsions
and sleepiness, coma. There is rapid weight loss and death a few months later from
malnutrition, heart failure, pneumonia or a parasitic infection. In the case of T.
brucei rhodesiense infections, there is no coma or nervous system symptoms as probably
patient dies before these can develop.
Recently on the increase, there are a minimum of 20,000 new cases a year;
50,000,000 people are at risk. Nagana prohibits cattle raising in a large area of Africa
causing further malnutrition.
|
|
|
AFRICAN TRYPANOSOMES
Structure of an African
Trypanosome
Trypanosomes are unicellular
protozoans (figure 3) with a single flagellum that contains microtubules
in the 9+2 arrangement typical of other flagella. At the base of the
flagellum is the kinetoplast (figure 3) which contains DNA in the form
of about 6000 catenated circles. The kinetoplast DNA is 10% of the total
cellular DNA and is the important
site of action of some anti-trypanosome drugs such as ethidium. The kinetoplast
is part of the single long
mitochondrion which changes morphology during various stages of life cycle.
Most other organelles are
those typical of any eucaryotic cell. At surface of the cell are
sub-membranous pellicular microtubules which give the trypanosome its
shape. These underlie a typical plasma membrane which is often
covered by an electron-dense surface coat (figure 3).
|
| |
|
Figure
3 |
 Epimastigotes grown in culture; kinetoplast (KP) is anterior to the
nucleus (N). In most species of Trypanosoma, this stage reproduces in the gut of the
vector. ©
Ohio State University, College of Biological Sciences
Epimastigotes grown in culture; kinetoplast (KP) is anterior to the
nucleus (N). In most species of Trypanosoma, this stage reproduces in the gut of the
vector. ©
Ohio State University, College of Biological Sciences
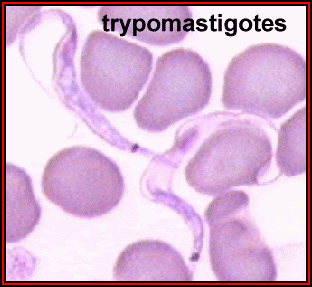
 Trypomastigotes in blood smear; kinetoplast is posterior to the
nucleus. This stage is found in all species of Trypanosoma, and in most species it is the
only stage that reproduces in the vertebrate (human) host. ©
Ohio State University, College of Biological Sciences
Trypomastigotes in blood smear; kinetoplast is posterior to the
nucleus. This stage is found in all species of Trypanosoma, and in most species it is the
only stage that reproduces in the vertebrate (human) host. ©
Ohio State University, College of Biological Sciences
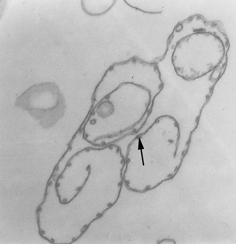
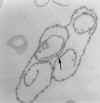 Pellicle membrane of a trypanosome. Left: The plasma membrane has been
ruptured to reveal the the underlying microtubules (arrow). Negative stain
electron micrograph. Right: Thin section electron micrograph showing
microtubules (arrow) and plasma membrane
Pellicle membrane of a trypanosome. Left: The plasma membrane has been
ruptured to reveal the the underlying microtubules (arrow). Negative stain
electron micrograph. Right: Thin section electron micrograph showing
microtubules (arrow) and plasma membrane
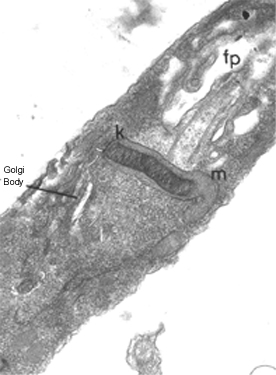
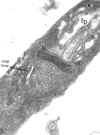 Thin section of a trypanosome (Leptomonas collosoma) showing the kinetoplast
(k) which is the DNA-containing region of the single mitochondrion (m). The
kinetoplast is found at the base of the flagellum and flagellar pocket (fp)
Thin section of a trypanosome (Leptomonas collosoma) showing the kinetoplast
(k) which is the DNA-containing region of the single mitochondrion (m). The
kinetoplast is found at the base of the flagellum and flagellar pocket (fp) |
|
Figure
4 |
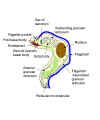 Diagram to show principal structures revealed by the electron microscope
in the bloodstream trypomastigote form of the salivarian trypanosome, Trypanosoma
congolense. It is shown cut in sagittal sections, except for most of the shaft of the
flagellum and the anterior extremity of the body. (Adapted from Vickerman, K., 1969. J
Protozool.
16:54-69.) (Note: the three sub-species (rhodesiense, gambiense and brucei)
cannot be distinguished morphologically)
Diagram to show principal structures revealed by the electron microscope
in the bloodstream trypomastigote form of the salivarian trypanosome, Trypanosoma
congolense. It is shown cut in sagittal sections, except for most of the shaft of the
flagellum and the anterior extremity of the body. (Adapted from Vickerman, K., 1969. J
Protozool.
16:54-69.) (Note: the three sub-species (rhodesiense, gambiense and brucei)
cannot be distinguished morphologically)
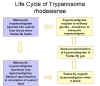 Life
cycle of Trypanosoma rhodesiense Life
cycle of Trypanosoma rhodesiense
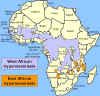 Distribution of West African or
Gambian Sleeping Sickness and East African or Rhodesian Sleeping Sickness Distribution of West African or
Gambian Sleeping Sickness and East African or Rhodesian Sleeping Sickness
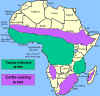 The distribution of tsetse fly and cattle raising areas
The distribution of tsetse fly and cattle raising areas
|
|
Figure
5 |
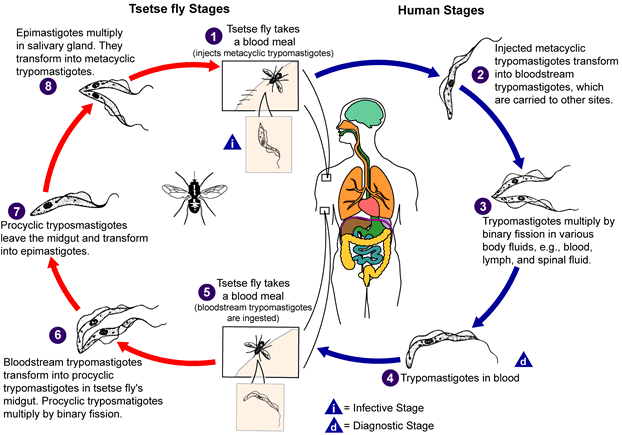
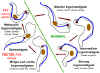 Life cycle of Trypanosoma
brucei. Developmental stages found in the bloodstream of the mammalian host, the midgut of
the tsetse, and the salivary glands of the tsetse. Redrawn from Vickerman
Life cycle of Trypanosoma
brucei. Developmental stages found in the bloodstream of the mammalian host, the midgut of
the tsetse, and the salivary glands of the tsetse. Redrawn from Vickerman
 During a blood meal on the mammalian host, an infected tsetse fly (genus
Glossina) injects metacyclic trypomastigotes into skin tissue.
The parasites enter the lymphatic system and pass into the bloodstream
During a blood meal on the mammalian host, an infected tsetse fly (genus
Glossina) injects metacyclic trypomastigotes into skin tissue.
The parasites enter the lymphatic system and pass into the bloodstream
 .
Inside the host, they transform into bloodstream trypomastigotes .
Inside the host, they transform into bloodstream trypomastigotes
 ,
are carried to other sites throughout the body, reach other blood fluids
(e.g., lymph, spinal fluid), and continue the replication by binary
fission ,
are carried to other sites throughout the body, reach other blood fluids
(e.g., lymph, spinal fluid), and continue the replication by binary
fission  . The
entire life cycle of African Trypanosomes is represented by
extracellular stages. The tsetse fly becomes infected with
bloodstream trypomastigotes when taking a blood meal on an infected
mammalian host ( . The
entire life cycle of African Trypanosomes is represented by
extracellular stages. The tsetse fly becomes infected with
bloodstream trypomastigotes when taking a blood meal on an infected
mammalian host ( , ,
 ).
In the fly’s midgut, the parasites transform into procyclic
trypomastigotes, multiply by binary fission ).
In the fly’s midgut, the parasites transform into procyclic
trypomastigotes, multiply by binary fission
 ,
leave the midgut, and transform into epimastigotes ,
leave the midgut, and transform into epimastigotes
 .
The epimastigotes reach the fly’s salivary glands and continue
multiplication by binary fission .
The epimastigotes reach the fly’s salivary glands and continue
multiplication by binary fission
 .
The cycle in the fly takes approximately 3 weeks. Humans are the
main reservoir for Trypanosoma brucei gambiense, but this species
can also be found in animals. Wild game animals are the main
reservoir of T. b. rhodesiense. .
The cycle in the fly takes approximately 3 weeks. Humans are the
main reservoir for Trypanosoma brucei gambiense, but this species
can also be found in animals. Wild game animals are the main
reservoir of T. b. rhodesiense.
DPDx, Division of
Parasitic Diseases CDC.
|
| |
Forms of T. Brucei in the mammalian
and insect hosts
The various stages of the life cycle of
T. brucei in each of its
hosts can be distinguished morphologically (figure 5)
|
 Figure
6 Glycolysis pathway in long-slender bloodstream forms of T.
brucei. The principal sites of inhibition by trypanocidal drugs in vivo are indicated by
red arrows. Abbreviations: SHAM, salicylhydroxamic acid; G-6-P, glucose-6-phosphate;
F-6-P, fructose-6-phosphate; FDP, fructose-I,6-diphosphate; GAP,
glyceraldehyde-3-phosphate; DHAP, dihydroxyacetone phosphate; GP sn-glycerol-3-phosphate;
diPGA, 1,3-diphosphoglycerate; 3PGA and 2PGA, 3- and 2-phosphoglycerate respectively; PEP,
phosphoenolpyruvate.
Figure
6 Glycolysis pathway in long-slender bloodstream forms of T.
brucei. The principal sites of inhibition by trypanocidal drugs in vivo are indicated by
red arrows. Abbreviations: SHAM, salicylhydroxamic acid; G-6-P, glucose-6-phosphate;
F-6-P, fructose-6-phosphate; FDP, fructose-I,6-diphosphate; GAP,
glyceraldehyde-3-phosphate; DHAP, dihydroxyacetone phosphate; GP sn-glycerol-3-phosphate;
diPGA, 1,3-diphosphoglycerate; 3PGA and 2PGA, 3- and 2-phosphoglycerate respectively; PEP,
phosphoenolpyruvate. |
Biochemistry and molecular biology of African trypanosomes
Oxidative metabolism
All living organisms make ATP as an energy carrier. This is produced
mainly by the oxidation of carbohydrates using glycolysis and the tricarboxylic acid
(TCA) cycle (figure 6). Because free living organisms (like us) do not have an abundance of food, we rely
on the much more efficient TCA cycle for most of our ATP production.
The trypanosome meets very different environments at different stages of
its life cycle. In the mammalian blood stream there is an abundance of oxygen and glucose.
The opposite is true in the insect gut or hemolymph. This is reflected in the number of
reactions of glycolysis and TCA cycle that can be carried out and in the elaboration of
the kinetoplast/mitochondrion. Clearly, if an organism can dispense with the TCA cycle it
can also dispense with its mitochondrion.
The forms of T. brucei in the insect gut have a full complement of
TCA and glycolysis enzymes. This is not surprising since nutrients are NOT abundant; in
most organisms that use oxidative phosphorylation, ATP production is sensitive to cyanide
because cytochromes a/a3 react with cyanide and can no longer transfer an
electron to oxygen to form water; however, oxidative phosphorylation in the insect gut
forms of Trypanosomes is only PARTIALLY CYANIDE SENSITIVE. Cytochrome a/a3
system is still CN--sensitive but in Trypanosomes there is an additional
cytochrome O which is CN- - insensitive. Little is known about the cytochrome
O system. In the insect, partial aerobic fermentation produces succinate, pyruvate, acetate
as well as carbon dioxide. Proline is a major fuel source.
The forms of T. brucei in the mammalian bloodstream use only
inefficient glycolysis because there is so much nutrient available; since glycolysis
produces much less ATP than the TCA cycle, respiration is 50 times that of normal
mammalian cell and in the bloodstream, T. brucei uses 10 times the amount of fuel
as in insect gut.
Note from biochemistry lectures. If you use only glycolysis to make ATP as
is done in anaerobic respiration in our muscles or in yeast, you cannot just
excrete pyruvate, which is what the trypanosome does in fact do, since you will quickly
reduce all of your NAD+ to NADH and the whole pathway will stop for lack of
oxidized substrate. We convert pyruvate to lactate while yeast converts it to ethanol to
oxidize our NADH back to NAD+ and keep glycolysis going. Trypanosomes excrete
pyruvate and have another way of converting NADH back to NAD+.
In trypanosomes, dihydroxyacetone phosphate metabolism is necessary for
reoxidation of NADH. This is an aerobic system, however, requiring oxygen but
oxygen consumption is CN- - insensitive so it does not use the usual cytochrome
chain. An FAD-containing dehydrogenase linked to copper containing oxidase. This complex
is the glycerophosphate oxidase system. Trypanosomes in the bloodstream depend on
this mechanism of keeping NAD oxidized. This system is the target of two trypanocidal
drugs: SURAMIN and SHAM (salicylhydroxamic acid) which is a chelating agent. Terminal
oxidase contains copper. These drugs have their specificity because this is a metabolic
pathway that mammals do not use.
There is another oddity in the trypanosomes' metabolism of carbohydrates.
Unlike mammalian cell, first nine reactions of glycolysis are organelle-associated
(GLYCOSOME). Why trypanosomes need to compartmentalize glycolysis is not clear. A
compartment does mean that all of the enzymes are concentrated in one place but diffusion
does not seem to be limiting in cells that do not have glycosomes.
|
| |
Kinetoplast morphology
The morphology of the
kinetoplast changes with metabolism carried out by the organism:
-
Bloodstream slender trypomastigoes: simple mitochondrion, few cristae are
short and tubular
-
Bloodstream short-stumpy: elaboration of mitochondrion, synthesis of
mitochondrial enzymes
-
Fly midgut: elaborate array of plate-like cristae. Mitochondrion extends
both anteriorally and posteriorally from kinetoplast
-
Fly metacyclic: degeneration of mitochondrion
|
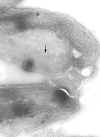
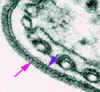 Figure 7
Electron micrograph of the surface coat of the mammalian form of a
trypanosome. The pink arrow indicates the coat and the blue arrow indicates the
underlying plasma membrane
Figure 7
Electron micrograph of the surface coat of the mammalian form of a
trypanosome. The pink arrow indicates the coat and the blue arrow indicates the
underlying plasma membrane
 Figure 8 Successive waves of parasites in the blood are characteristic
of sleeping sickness and correlate with cycles of fever. A population of
parasites (with a few variable surface glycoproteins (VSGs)) divides in the
bloodstream over a period of days. Some
of these trypanosomes have VSG A on their surface (clone A). The immune system raises antibodies against
all of the population's antigens and, as a result, most of the parasites die. A few
trypanosomes, however, change their coat so that they express another VSG
(e.g. VSV B). These parasites survive by expressing the new VSG gene and give rise to a new population
(clone B). In time, the host raises antibodies against VSG B and clone B cells
die off but again a few cells change their coats and survive. This cycle is repeated many times in the course of a
chronic infection as parasites keep expressing new genes and displaying new VSG
antigens. From
each successive population it is possible to isolate individual trypanosomes and from them
to grow clones expressing particular VSGs.
Figure 8 Successive waves of parasites in the blood are characteristic
of sleeping sickness and correlate with cycles of fever. A population of
parasites (with a few variable surface glycoproteins (VSGs)) divides in the
bloodstream over a period of days. Some
of these trypanosomes have VSG A on their surface (clone A). The immune system raises antibodies against
all of the population's antigens and, as a result, most of the parasites die. A few
trypanosomes, however, change their coat so that they express another VSG
(e.g. VSV B). These parasites survive by expressing the new VSG gene and give rise to a new population
(clone B). In time, the host raises antibodies against VSG B and clone B cells
die off but again a few cells change their coats and survive. This cycle is repeated many times in the course of a
chronic infection as parasites keep expressing new genes and displaying new VSG
antigens. From
each successive population it is possible to isolate individual trypanosomes and from them
to grow clones expressing particular VSGs.
|
Host reactions to the
parasite
Any parasite faces a problem when it takes up residence within another
organism. The latter is likely to mount an immune response to the parasite which may
destroy it. Some parasites hide behind surfaces that disguise them so that to the immune
system they look like normal cells. Other parasites (including Trypansoma cruzi)
enter cells to get away from the immune system. T. brucei does neither. Instead,
The parasite is very antigenic which is one reason for the symptoms shown
by the infected patient. But the number of parasites in the bloodstream does not go on
increasing and increasing until the patient dies. The patient undergoes waves of fever and
cycles in parasite infestation. The waves of parasitemia correlate with the fever
observed. Number of parasites in blood shows waves as the immune system partially
overcomes the infection. Cyclic nature of parasitemia is very characteristic.
So why does not the massive immune response mounted by the infected
individual clear the body of the parasite as it ought to do? Although massive immune
response with strikingly high levels of Ig (especially IgM) and profound B lymphocyte
proliferation, it must be that there is a change effected in some trypanosomes of the
total population that allows them to seed a new generation of parasites. Later in the
chronic phase of the infection, lymphoid organs are depleted of lymphocytes, they shrink
and patchy fibrosis replaces the lymphocytes. Immunodepression sets in and the parasitemia
is uncontrolled leading to death.
Why are not all of the parasite destroyed by the massive immune response?
This is the key to the progress of the disease! The answer must lie in the electron-dense
surface coat (figure 7) that surrounds the bloodstream forms of the parasite and which is the only
major antigen recognized by the host=s immune
system.
If we clone single cells from different infected animals or patients, the
coat is biochemically different! Not just a bit different but so different that the coat
protein must come from the expression of different genes by trypanosomes in each animal.
Moreover, if we take cells from a defined wave of parasitemia in the same patient,
it is found that all of the trypanosomes in that wave of organisms are expressing the
same single surface antigen whereas in other waves, all of the parasites are
expressing a single but completely different antigen (figure 8). In other words, a different
surface antigen gene is being expressed. The surface coast is therefore made of VARIABLE
SURFACE ANTIGENS or VARIABLE SURFACE GLYCOPROTEINS (VSGs). You will also see
the term VATs for variable antigen types.
Thus escape from the immune response depends upon the ability to express a
new VSG. Since hundreds of these waves of parasitemia can occur before the host dies (in a
laboratory situation, normal number of waves is much fewer that this) and no antigen is
repeated, there must be an equal number of VSG genes. In fact, there are probably
1000-2000 such genes C10% of the cell=s genome is devoted to genes that express these
surface molecules that allow the organism to be one step ahead of the host's immune
response.
This leads to a series of questions: What is the structure of these VSGs?
How is the switch effected? How is only one VSG gene is expressed at a time? How is
complete release of the VSG carried out?
Each VSG glycoprotein has a size of about 65kD, about 500 amino
acids and has three domains. At the N-terminus is the signal sequence;
the next 360 amino acids are usually very different
from the similar sequence in other VSGs. The 120 C-terminal amino acids are quite similar in different VSGs. This latter
part is hidden from the immune system by being next to the plasma membrane.
From protein sequencing and cDNA sequencing we get a different picture of
the C-terminal part of the molecule. The cDNA (gives sequence encoded by gene)
shows a typical transmembrane hydrophobic sequence that is used normally to attach the
protein to the plasma membrane together with a short intracellular domain but protein
sequencing shows that the transmembrane part of the protein and the intracellular part
are no there in the mature protein. They are replaced by a weird structure that contains
sugars, ethanolamine, phospho-inositol and fatty acids. This structure is common to all
VSGs and is highly antigenic when purified but not in vivo. This suggests that the
VSGs in the coat are tightly packed to exclude antibodies. Therefore this site is of nor
use in vaccine development.
|
 Figure 9 When an African trypansome expresses a
new coat, this is frqquently associated with the appearance ofg a new
copy of the gene for that coat. This is called the expression- linked
copy
Figure 9 When an African trypansome expresses a
new coat, this is frqquently associated with the appearance ofg a new
copy of the gene for that coat. This is called the expression- linked
copy |
How does the sequential
expression of VSGs occur?
Restriction endonuclease digestion, gel electrophoresis and Southern
blotting allows us to fragment the trypanosomes=
genome and separate the fragments. We can now detect a particular gene in the genome
displayed on our gel by hybridizing it with a complementary probe. This is usually cDNA
made against mRNA or against a cloned gene.
If we are lucky with our fragmentation and there is only one gene in the
genome for a particular protein, we should see only one band that will hybridize on the
gel (providing that an endonuclease has not cleaved within the gene). Such analyses showed
that each VSG gene was normally expressed only once in the genome which is not surprising
since VSG genes constitute so much of the genome and multiple copies would occupy a very
large proportion of the genome.
We can take DNA from organisms in the first, second, third and
fourth waves of parasitemia (clones A, B, C and D). We can then probe
these DNAs after fragmentation with a probe that detects the gene expressed in
second wave, that is in clone B. We find that clones A, C and D (which are not
expressing VSG B) have only one copy of the VSG B.
However, a very surprising result was found, as shown in the
diagram at the left (figure 9), when we probe for the gene that codes for VSG B in cells that are
expressing VSG B.
|
| |
There is an extra copy of the gene that is being expressed in
this particular clone of trypanosomes. IT IS ONLY THE GENE THAT IS BEING EXPRESSED THAT
OCCURS IN AN EXTRA COPY! THUS WE HAVE GENE DUPLICATION but it is only temporary as
when the trypanosome switches to a new VSG the extra copy usually disappears and is
replaced by another EXPRESSION-LINKED COPY (ELC). It is the extra copy that is being
transcribed into mRNA for translation into protein, not the copy that is permanently in
genome. The new copy is in an EXPRESSION SITE. Expression sites are always close to a
TELOMERE, that is near the end of a chromosome. An ELC and a permanent gene may be on
different chromosome. This suggests a copy/translocate
mechanism for a cassette of information. Sometimes, an expression linked copy is not
produced bu the permanent copy is transcribed. These transcribed non-ELC genes are always
at telomeres. The temporary copying of a gene and using the copy is very unusual.
Can the genes be expressed in any order? Is there a preprogrammed sequence
of expression? One would suspect not since there are thousands of VSG genes and there
would be a selective advantage of being able to use all of them. Indeed, the order is not
absolute. At the beginning of the infective phase, VSGs are produced by parasites in the
insect salivary glands. A subset of the repertoire (about 12) of the VSGs are produced
here. This remains during the first wave of parasitemia in the mammal. The whole
repertoire is then open to expression and there is preferred but not fixed order of
expression. Each gene can be expressed only once during an infection. Note that the fact
that there is an initial subset of VSGs that are expressed gives hope for a vaccine.
mRNAs in trypanosome are also very unusual. Almost every mRNA that has
been looked at starts with the same 35 nucleotides. This is coded by an exon far away from
the rest of the gene.
Shedding of the VSG coat
It will clearly be important that the parasite sheds
all of its coat
at a given time in order to survive the developing immune response against its original
VSG. We saw earlier that the form of the VSG at the cell surface is attached through a
glycolipid and not via a transmembrane protein sequence. This may give a clue to how
shedding occurs. A trypanosome- and VSG-specific phospholipase C is found in bloodstream
forms. Thus since all VSGs have the same attachment structure only one enzyme is needed
for rapid and complete cleavage.
Possibilities for
chemotherapy
The existence of possibly 2000 VSG genes makes a wide spectrum vaccine
very unlikely. Unfortunately, the common antigens are buried away from the immune
response. Perhaps an agent interacting with the 35 nucleotide sequence of the mRNAs that
are found in all trypanosomes but in no host animals mRNAs could be effective but none is
known. The best possibility is a vaccine against the 12 or so initial antigens that are
expressed in the organisms that are injected by the tsetse fly. Alternatively, an
inhibitor of the phospholipase C might be a very good candidate.
|
| |
Extraordinary RNA editing by
trypanosomes: The mystery of the missing genes
These organisms have proved to be biochemical oddities in many respects
but when some of the findings in trypanosomes have been looked for in other species, these
apparently unique biochemical processes have turned out not to be unique after all.
The trypanosomes have a very odd nuclear genome. The chromosomes do not
condense in nuclear division and so we do not know how many chromosomes there are. The
nuclear genes include 1000-2000 genes that encode the variable surface antigens that allow
the coast of the organisms to be changed regularly so that it can avoid the host=s immune response. The way in which this is done is
extraordinary and involves the shifting of a new copy of a gene into an expression site
when it is needed. Up to 10% of the genome is composed of all of these genes for variable
surface antigens.
In addition, there is the kinetoplast. This DNA-rich structure lies at the
base of the flagellum and is at one end of the single long mitochondrion of the
flagellate. It is equivalent to the mitochondrial DNA of all other cells but makes up a
very much greater proportion of the DNA of the cell than does the single circle
mitochondrial DNA of other cells. Remember that our mitochondria can code for a few of
their own proteins (some cytochrome subunits and ribosome subunits) together with all of
the mitochondrial ribosomal RNAs and all of the mitochondrial transfer RNAs. Although
the kinetoplast DNA is much more elaborate and is a greater proportion of the cell's DNA,
it does not code for any more RNAs or proteins than other mitochondrial DNAs. Indeed, some
of the tRNAs of the kinetoplast are not encoded in this DNA and have to be imported from
the cytoplasm which is not the case with mammalian mitochondria. The mechanism for this
import is unknown.
The reason that kinetoplast DNA makes up such a high proportion of the
total DNA of the cells is it complexity.
It contains 20-50 copies of a 22kb MAXI CIRCLE that is
equivalent to mitochondrial DNA in any other mitochondrion. BUT in addition, there are up
to 10,0000 1kb MINI-CIRCLES of , until recently, unknown function. These are odd in
another way: they form a single network of catenated circles.
As already noted, among the things coded for by the maxi circle DNA are
ribosomal RNA of mitochondrion and a variety of the enzymes of the mitochondria
respiratory chain. There are also several unidentified open reading frames in the
maxicircle DNA.
|
| |
The enigma of the cytochrome
oxidase of some trypanosomes, especially Trypansosma brucei
In all of the trypanosomes that have been looked at, the cytochrome
oxidase subunit III (CO III) is encoded in the kinetoplast DNA. All, that is, except
T.
brucei. Not only do all but T. brucei have their COIII genes in the kinetoplast
DNA, but they are all in the same position within that DNA. Yet with the exception
of the COIII gene, T. brucei kDNA looks very like that of two other trypanosomes,
Crithidia
fasciculata and Leishmania tarentolae.
The only real difference is the lack of COIII gene. In the other two
species, this gene is upstream of the apocytochrome b gene. Does this mean that
T.
brucei has no cytochrome oxidase subunit III? No it must have for it has to carry out
oxidative phosphorylation. In addition, the mRNA and the protein are clearly in the
organism. Perhaps the gene is just not in the same place as the similar gene in the other
trypanosomes. But we cannot find it elsewhere. Probing the DNA either of the nucleus or
the kinetoplast shows that there is no gene. How can we have a protein and an mRNA without
a gene to code for it?
Since in other trypanosomes that clearly have the COIII gene, it is always
in the same place, perhaps we should look again at this region of the kinetoplast genome!
So what is there in this region of kDNA in T. brucei? There are a few open reading
frames that might code for small proteins but nothing of the size of the COIII protein
and, in any case, the two largest have no start AUG codon so cannot code for a real
protein. Even if they did, they would code for a very unusual protein which would
be very rich in charged amino acids for the DNA in this region is very G-C rich.
As already noted, investigators have looked for the COIII gene elsewhere
in the kDNA maxi circle but not found it. The kDNA maxi and mini circles have now been
sequenced. Using probes that will detect the COIII gene in L. tarentolae and
C.
fasciculata, we do not find any apparent COIII gene in maxicircle or the nuclear DNA
of T. brucei.
This means that the COIII gene is either missing OR highly diverged so
that probes do not pick it up. But, again as noted above, it cannot be missing as the
T.
brucei has a cyanide-sensitive cytochrome system similar to the other two. It would
also not be expected to be highly diverged in view of the extremely high conservation of
the COIII genes in all other trypanosomes that have been looked at.
If you cannot find the gene, one can look for the transcripts, i.e. the
mRNAs.
Evidence was found for an mRNA transcript in
T. brucei that had
a sequence similar to the gene for COIII in the other trypanosomes. The similarity of
the sequences allows the determination of the correct open reading frame for the
T.
brucei transcript.
Of the 181 amino acids predicted by the sequence, 135 conserved amongst
all three. If we take conservative replacements and those conserved in one other species
we find 160 out of 181 are conserved. This level of conservation (88%) is slightly better
than that between T. brucei and L. tarentolae maxi circles genes where the
conservation ranges from 65% to 84%.
|
| |
This means that there must
be a gene for the COIII protein in T. brucei but where is it?
Again we must ask: Is there a genomic sequence that matches the COIII
transcript?
Before it has been shown that heterologous hybridization using L.
tarentolae and C. fasciculata COIII probes could not detect COIII sequences in
T.
brucei genes. This was confirmed by Southern blot analyses using probes that were
predicted form the sequence that was obtained for the transcript. The probes do not
hybridize to kDNA or to total DNA.
No detection could be found with probes that would have detected 0.5
copies of the gene per genome.
Since there is no gene apparently for these transcripts in
T. brucei,
two possibilities:
1. The transcript may be made by splicing together small fragments of RNA
transcribed form multiple sites. There is after all precedent for mini-exons in the
trypanosome system. There are data that would tend to exclude this possibility.
2. There is severe editing of the transcript after or during
transcription sot that the final transcript is nothing like the gene from which it was
transcribed.
So now we must go back to the kinetoplast genome. The gene MUST be there,
we just cannot detect it
and the place to look for the gene for COIII would be immediately upstream
of the apo-cytochrome b gene where the COIII gene is located in so many other
trypanosomes.
The sequence in this position which we have seen does not contain large
open reading frames with start codons was reinvestigated. But, in fact, the sequence of
this region matches the transcript sequence exactly EXCEPT for the presence of uridines in
the transcript that are not in the DNA of the gene. Note: start codons are ATG so that
explains the lack of start codons in the gene. The lack of 25% of the nucleotides explains
the smaller open reading frames. This also explains the high G-C content.
In spite of the differences between the RNA and DNA sequences, only the
number and positions of Us are affected; Cs, Gs, and As occur in the same sequence in the
mRNA and the genomic DNA.
Of the 626 nucleotides sequenced initially in the
T. brucei COIII
transcripts, 347 are URIDINES that are not coded for by the gene. They are added in 121
different sites.
At one of those sites, the addition of a U creates a stop codon exactly
where the native stop codon occurs in the other COIII genes.
In addition, 16 uridines, predicted by the genomic sequence at 7 sites
appear to be deleted.
The protein coding portion of the transcript contains 315 additions and 15
deletions in 546 nucleotides.
Thus 58% of the coding part of the mRNA results from editing and is not in
the original gene.
The editing is the reason why cDNA probes do no hybridize to the genomic
DNA.
Why do this?
One suggestion for this editing is that it might serve as a control
mechanism during the complex lifecycle of the parasite. Of course, not using Us in the
gene saves space--or does it?
How does the editing take place? What determines where the Us are put in
and taken out? Is there an original transcript that corresponds to the kinetoplast genomic
sequence?
The first step is indeed to make a transcript that is complementary to the
genomic sequence. This is now edited by putting in or taking out uridines at specific
places. The sequence of a nucleic acid is usually determined by another nucleic acid and a
whole new class of RNAs that are at present unique to the trypanosomes are used for this.
These small RNAs are called guide RNAs (gRNAs) are coded for by regions of the maxi-circle
and of the mini-circle for which previously no function was known.
One gRNA has a 5' end that is complementary to a short region of the
unedited COIII initial mRNA transcript (near the 3' end). At the end of this region of the
gRNA there will be one or more adenosines that are not in the primary transcript followed
by more sequence and a poly U tail. At the site of non-complementarity the primary
transcript is cut by an endonuclease and a U (from the poly U tail) is transferred on (by
terminal U transferase). This is now complementary to the A in the gRNA. If there is
another A in the gRNA, another U will be added to the primary transcript and this will go
on till there is not another A in the sequence. This is the signal for a ligase enzyme to
attach the original 5' end of the primary transcript to the newly inserted U (or Us) on
the 5' end of the 3' portion of the primary transcript. Presumably now the gRNA
dissociates. But now a new sequence containing Us is present in the initially edited
primary transcript and this is recognized by a different gRNA which can edit further
towards the 5' end of the primary transcript. Thus there is sequential editing from the 3'
to the 5' end of the original mRNA transcript and intermediates in processing have been
found that support this idea. Whatever, the mechanism, we have an extraordinary situation
in which most of the uridines of a gene are left out and are put in after transcription to
mRNA using the coding potential of another set of genes that code of the gRNAs. This seems
very inefficient and has only been found in trypanosomes to this extent.
|
 Figure 10 Vector: Triatoma infestans (assassin bugs) and related species and
genera (e.g., Rhodnius and Panstrongylus) © Ohio State University, College of Biological Sciences
Figure 10 Vector: Triatoma infestans (assassin bugs) and related species and
genera (e.g., Rhodnius and Panstrongylus) © Ohio State University, College of Biological Sciences
|
AMERICAN TRYPANOSOMES
Trypanosoma cruzi
Chagas Disease
The disease is carried by reduvid bugs
including the assasin bugs and rhodnius (figure 10) which infect the patient
when they defecate after taking a blood meal
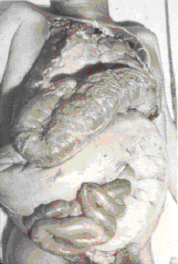
The symptoms of Chagas' disease are:
chronic infection, neurological disorders (including dementia),
megacolon (figure 11), megaesophagus, and damage to the heart muscle (figure
11). Chagas' disease is often fatal
unless treated.
In acute disease, there is
often severe anemia, muscle pain and neurological
disorders. The latter are common in children under 2 years in which death may occur in about
a month. Chronic disease may be mild and sometimes asymptomatic but there may be
damage to nerves causing cessation of gut muscle contractions, irregular
heartbeat and destruction of nervous system motor centers. The chronic
form of the disease is found in adults but most likely
arises from a childhood infection. T. cruzi can cross the placenta and so
chronically-infected mothers can infect their babies which may succumb to the very
acute form of the disease
|
|
Figure 11 |
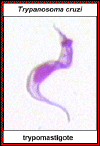 Trypomastigote of T. cruzi ©
Ohio State University, College of Biological Sciences
Trypomastigote of T. cruzi ©
Ohio State University, College of Biological Sciences
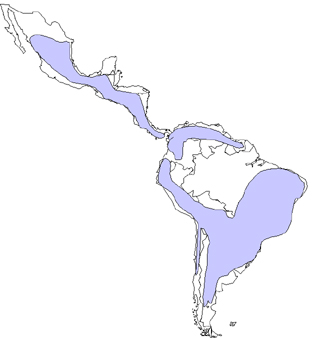
 Worldwide distribution of Chaga's disease
Worldwide distribution of Chaga's disease
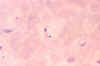 Trypanosoma cruzi in blood smear. CDC
Trypanosoma cruzi in blood smear. CDC
 Amastigotes (pseudocyst) of T. cruzi in the heart of a dog. ©
Ohio State University, College of Biological Sciences
Amastigotes (pseudocyst) of T. cruzi in the heart of a dog. ©
Ohio State University, College of Biological Sciences
|
| |
 This child from Panama is suffering from Chagas disease manifested as an acute infection with swelling of the right eye
This child from Panama is suffering from Chagas disease manifested as an acute infection with swelling of the right eye
 Trypanosoma cruzi in monkey heart. CDC/Dr. L.L. Moore, Jr. 1969
Trypanosoma cruzi in monkey heart. CDC/Dr. L.L. Moore, Jr. 1969
 Life cycle of Trypanosoma cruzi
Life cycle of Trypanosoma cruzi
 Megacolon in a Chagas' disease patient
Megacolon in a Chagas' disease patient
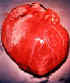 Dilated cardiomegaly caused by T. cruzi
(D. Despommier
from
Parasites on the web)
Dilated cardiomegaly caused by T. cruzi
(D. Despommier
from
Parasites on the web)
|
|
WEB RESOURCES
Division
of Parasitic Diseases - Centers for Disease Control
|
Like, the African trypanosomes,
T. cruzi escapes from host immune
response but it does not do so by changing its antigenic coat. Thus, there is no
antigenic variation. Instead, it
escapes by hiding inside cells. The disease starts after a bite by the insect
vector. After 7-14 days the trypanosomes arrive in the lymph nodes where they
divide. Here, they form aggregates called pseudocysts.. When the pseudocysts
rupture, the released parasites can enter cells in various parts of the body
including lymphatic tissue, muscle and tissue around nerve ganglia. The invasion
of the cardiac nerve ganglia is the cause of much of the heart disease in areas
where T. Cruzi is found.
Binding to and infection of the host cell
Binding
Amastigotes bind to the cell surface (e.g. a monocyte) via a variety of
receptor proteins including fibronectin. Sialic acid is important as cells deficient in sialic acid are not penetrated.
Interestingly, T. cruzi has an enzyme called trans-sialidase which actually puts more sialic
acid onto cells, thereby enhancing uptake.
Entry into the cell
Uptake is by a process of induced phagocytosis.
Lysosomes migrate to the cell surface when cells come in contact with T. cruzi
and the parasite enters a cytoplasmic vacuole, the parasitophorous
vacuole, that is formed from a lysosome. Thus, agents that induce the migration of lysosomes from peri-nuclear areas
so that they underlie the plasma membrane, enhance infectivity. Conversely, blocking lysosomal function stops infection.
Somehow, the parasites escape from the destructive potential of the lysosome
and after about an hour T. cruzi releases a protein toxin that inserts into
the membrane at the low pH typical of this organelle. As a result T. cruzi
escapes into the cytoplasm as the lysosomal membrane is destroyed.
|
| |
| |
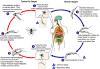
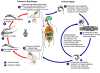 An infected triatomine insect vector (or “kissing”
bug) takes a blood meal and releases trypomastigotes in its feces near
the site of the bite wound. Trypomastigotes enter the host through
the wound or through intact mucosal membranes, such as the conjunctiva
.
Common triatomine vector species for trypanosomiasis belong to the
genera Triatoma, Rhodinius, and Panstrongylus.
Inside the host, the trypomastigotes invade cells, where they
differentiate into intracellular amastigotes
.
The amastigotes multiply by binary fission
and differentiate into trypomastigotes, and then are released into the
circulation as bloodstream trypomastigotes
.
Trypomastigotes infect cells from a variety of tissues and transform
into intracellular amastigotes in new infection sites. Clinical
manifestations can result from this infective cycle. The
bloodstream trypomastigotes do not replicate (different from the African
trypanosomes). Replication resumes only when the parasites enter
another cell or are ingested by another vector. The “kissing”
bug becomes infected by feeding on human or animal blood that contains
circulating parasites
.
The ingested trypomastigotes transform into epimastigotes in the
vector’s midgut .
The parasites multiply and differentiate in the midgut
and differentiate into infective metacyclic trypomastigotes in the
hindgut .
An infected triatomine insect vector (or “kissing”
bug) takes a blood meal and releases trypomastigotes in its feces near
the site of the bite wound. Trypomastigotes enter the host through
the wound or through intact mucosal membranes, such as the conjunctiva
.
Common triatomine vector species for trypanosomiasis belong to the
genera Triatoma, Rhodinius, and Panstrongylus.
Inside the host, the trypomastigotes invade cells, where they
differentiate into intracellular amastigotes
.
The amastigotes multiply by binary fission
and differentiate into trypomastigotes, and then are released into the
circulation as bloodstream trypomastigotes
.
Trypomastigotes infect cells from a variety of tissues and transform
into intracellular amastigotes in new infection sites. Clinical
manifestations can result from this infective cycle. The
bloodstream trypomastigotes do not replicate (different from the African
trypanosomes). Replication resumes only when the parasites enter
another cell or are ingested by another vector. The “kissing”
bug becomes infected by feeding on human or animal blood that contains
circulating parasites
.
The ingested trypomastigotes transform into epimastigotes in the
vector’s midgut .
The parasites multiply and differentiate in the midgut
and differentiate into infective metacyclic trypomastigotes in the
hindgut .
Trypanosoma cruzi can also be transmitted through blood
transfusions, organ transplantation, transplacentally, and in laboratory
accidents.
DPDx,
Division of Parasitic Diseases CDC.
|
|
|
 Return to the Parasitology section of Microbiology and Immunology On-line
Return to the Parasitology section of Microbiology and Immunology On-line
This page last changed on
Tuesday, February 17, 2015
Page maintained by
Richard Hunt
|
 Figure 1. Tsetse fly: the vector for African trypansomiasis ©
Ohio State University, College of Biological Sciences
Figure 1. Tsetse fly: the vector for African trypansomiasis ©
Ohio State University, College of Biological Sciences
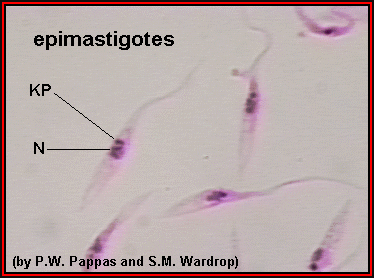 Epimastigotes grown in culture; kinetoplast (KP) is anterior to the
nucleus (N). In most species of Trypanosoma, this stage reproduces in the gut of the
vector. ©
Ohio State University, College of Biological Sciences
Epimastigotes grown in culture; kinetoplast (KP) is anterior to the
nucleus (N). In most species of Trypanosoma, this stage reproduces in the gut of the
vector. ©
Ohio State University, College of Biological Sciences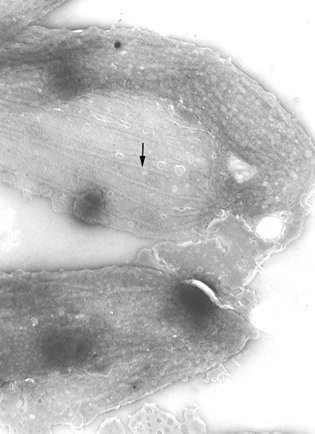
 Pellicle membrane of a trypanosome. Left: The plasma membrane has been
ruptured to reveal the the underlying microtubules (arrow). Negative stain
electron micrograph. Right: Thin section electron micrograph showing
microtubules (arrow) and plasma membrane
Pellicle membrane of a trypanosome. Left: The plasma membrane has been
ruptured to reveal the the underlying microtubules (arrow). Negative stain
electron micrograph. Right: Thin section electron micrograph showing
microtubules (arrow) and plasma membrane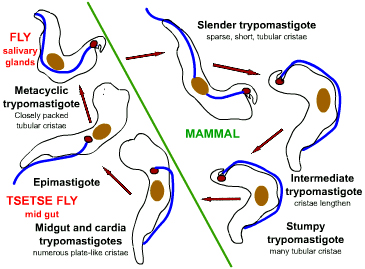 Life cycle of Trypanosoma
brucei. Developmental stages found in the bloodstream of the mammalian host, the midgut of
the tsetse, and the salivary glands of the tsetse. Redrawn from Vickerman
Life cycle of Trypanosoma
brucei. Developmental stages found in the bloodstream of the mammalian host, the midgut of
the tsetse, and the salivary glands of the tsetse. Redrawn from Vickerman
 Figure
6 Glycolysis pathway in long-slender bloodstream forms of T.
brucei. The principal sites of inhibition by trypanocidal drugs in vivo are indicated by
red arrows. Abbreviations: SHAM, salicylhydroxamic acid; G-6-P, glucose-6-phosphate;
F-6-P, fructose-6-phosphate; FDP, fructose-I,6-diphosphate; GAP,
glyceraldehyde-3-phosphate; DHAP, dihydroxyacetone phosphate; GP sn-glycerol-3-phosphate;
diPGA, 1,3-diphosphoglycerate; 3PGA and 2PGA, 3- and 2-phosphoglycerate respectively; PEP,
phosphoenolpyruvate.
Figure
6 Glycolysis pathway in long-slender bloodstream forms of T.
brucei. The principal sites of inhibition by trypanocidal drugs in vivo are indicated by
red arrows. Abbreviations: SHAM, salicylhydroxamic acid; G-6-P, glucose-6-phosphate;
F-6-P, fructose-6-phosphate; FDP, fructose-I,6-diphosphate; GAP,
glyceraldehyde-3-phosphate; DHAP, dihydroxyacetone phosphate; GP sn-glycerol-3-phosphate;
diPGA, 1,3-diphosphoglycerate; 3PGA and 2PGA, 3- and 2-phosphoglycerate respectively; PEP,
phosphoenolpyruvate.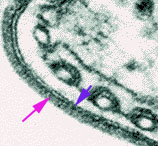 Figure 7
Electron micrograph of the surface coat of the mammalian form of a
trypanosome. The pink arrow indicates the coat and the blue arrow indicates the
underlying plasma membrane
Figure 7
Electron micrograph of the surface coat of the mammalian form of a
trypanosome. The pink arrow indicates the coat and the blue arrow indicates the
underlying plasma membrane
 Figure 9 When an African trypansome expresses a
new coat, this is frqquently associated with the appearance ofg a new
copy of the gene for that coat. This is called the expression- linked
copy
Figure 9 When an African trypansome expresses a
new coat, this is frqquently associated with the appearance ofg a new
copy of the gene for that coat. This is called the expression- linked
copy  Figure 10 Vector: Triatoma infestans (assassin bugs) and related species and
genera (e.g., Rhodnius and Panstrongylus) © Ohio State University, College of Biological Sciences
Figure 10 Vector: Triatoma infestans (assassin bugs) and related species and
genera (e.g., Rhodnius and Panstrongylus) © Ohio State University, College of Biological Sciences
 Trypomastigote of T. cruzi ©
Ohio State University, College of Biological Sciences
Trypomastigote of T. cruzi ©
Ohio State University, College of Biological Sciences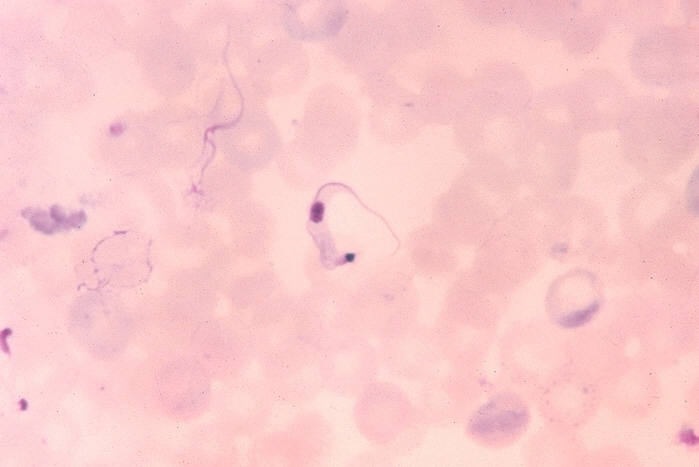 Trypanosoma cruzi in blood smear. CDC
Trypanosoma cruzi in blood smear. CDC
 This child from Panama is suffering from Chagas disease manifested as an acute infection with swelling of the right eye
This child from Panama is suffering from Chagas disease manifested as an acute infection with swelling of the right eye Life cycle of Trypanosoma cruzi
Life cycle of Trypanosoma cruzi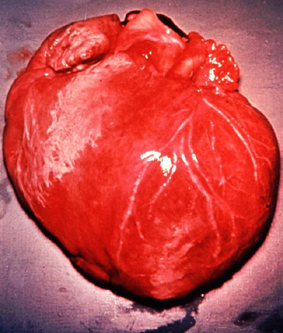 Dilated cardiomegaly caused by T. cruzi
(D. Despommier
from
Parasites on the web)
Dilated cardiomegaly caused by T. cruzi
(D. Despommier
from
Parasites on the web)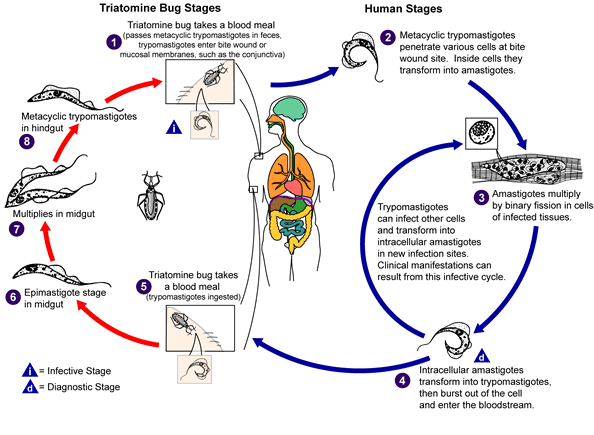 An infected triatomine insect vector (or “kissing”
bug) takes a blood meal and releases trypomastigotes in its feces near
the site of the bite wound. Trypomastigotes enter the host through
the wound or through intact mucosal membranes, such as the conjunctiva
An infected triatomine insect vector (or “kissing”
bug) takes a blood meal and releases trypomastigotes in its feces near
the site of the bite wound. Trypomastigotes enter the host through
the wound or through intact mucosal membranes, such as the conjunctiva