|
x |
x |
|
 |
 |
|
DOENÇAS
INFECCIOSAS |
BACTERIOLOGIA |
IMUNOLOGIA |
MICOLOGIA |
PARASITOLOGIA |
VIROLOGIA |
|
|
PARASITOLOGIA - CAPÍTULO TRÊS
PARASITOLOGIA MOLECULAR: TRIPANOSSOMAS
CÉLULAS EUCARIÓTICAS COM UM JEITO DIFERENTE DE FAZER AS COISAS
Dr
Richard Hunt
Emeritus Professor
University of South Carolina School of Medicine
Tradução: PhD. Myres Hopkins
|
|
EM INGLÊS
|
|
SHQIP - ALBANIAN |
Dê a
sua opinião
CONTATO |
|
SEARCH |
E-MAIL
DR MYRES HOPKINS |
|
ESCOLA DE MEDICINA DA
UNIVERSIDADE DA CAROLINA DO SUL |
|
|

 |
|
Logo image © Jeffrey
Nelson, Rush University, Chicago, Illinois and
The MicrobeLibrary |
|
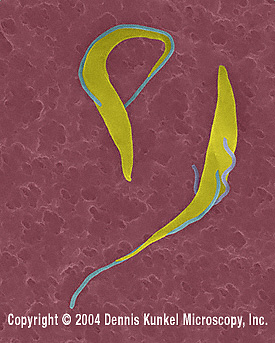
Trypanosome
trypomastigotes (Trypanosoma spp.).
©
Dennis Kunkel Microscopy, Inc.
Used with permission
 |
Nesta seção veremos a
doença tripanossomíase e o ciclo de vida dos parasitas que a provocam. Os
aspectos interessantes do ciclo de vida nos conduzem às maneiras pelas quais o
parasita adquiriu vias bioquímicas não usuais para se adaptar ao seu nicho.
Estas oferecem alternativas potenciais para a quimioterapia.
Tripanossomas são parasitas
bem sucedidos que conseguem escapar da resposta imune do hospedeiro; isto
acontece através de um mecanismo muito complexo de mudança de antígenos e é o
conhecimento deste mecanismo que nos guia aos primeiros passos para o
desenvolvimento de uma vacina anti-tripanossoma.
TRIPANOSSOMAS
Tripanossomas pertencem à ordem KINETOPLASTIDA, assim chamada devido à grande
estrutura que contém DNA, o cinetoplasto, encontrada na base do flagelo. |
| |
|
DOENÇAS CAUSADAS PELOS TRIPANOSSOMAS |
|
Espécies de tripanossoma |
|
Espécies |
Distribuição |
Espécies que exibem a doença |
Vetor |
Doença |
Tratamento |
| Trypanosoma brucei gambiense
|
Oeste da África |
humanos |
Glossina
palpalis (Môsca tse-tse)

© OhioState University,
College of Biological Sciences
|
Tripanossomíase (Doença do sono)
crônica
|
 Sinal de Winterbottoms
(CDC)
Sinal de Winterbottoms
(CDC) |
Suramin, pentamidina e berenil: podem curar a infecção se
administrados antes da invasão do sistema nervoso.
Difluorometilornitina (DMFO) mesmo se o parasita invadiu o
cérebro. |
|
Trypanosoma brucei rhodesiense |
Leste da África |
humanos |
Glossina morsitans |
Tripanossomíase (Doença do sono)
aguda |
 Paciente com doença do sono avançada
(CDC)
Paciente com doença do sono avançada
(CDC) |
Idem |
| Trypanosoma brucei brucei |
África |
Gado, antílope, cavalos, camelos |
Glossina pallidipes |
Nagana
aguda |
 Ordenhando com nagana
© WHO
Ordenhando com nagana
© WHO |
|
| Trypanosoma congolense |
África |
Gado, antílope, cavalos, camelos |
Glossina morsitans |
Nagana
crônica |
|
|
| Trypanosoma vivax |
África |
Gado, antílope, cavalos, camelos |
Glossina morsitans |
Nagana
aguda |
|
|
| Trypanosoma equiperdum |
África |
Cavalos, jumentos |
Nenhum (transmitida durante o coito) |
Dourine
aguda |
|
|
| Trypanosoma evansi |
África |
Camelos, cavalos, veado |
Môsca Tabanid

From Rob
Hutchinson
|
Sura |
|
|
| Trypanosoma equinum |
|
Camelos,
cavalos, veado |
Môsca Tabanid |
Mal de Caderas |
|
|
| Trypanosoma cruzi |
América do Sul e Central |
|
Reduvídeos: Rhodnius prolixus

© OhioState University,
College of Biological Sciences
|
Doença de Chagas |
|
Não há tratamento eficiente. As drogas disponíveis apenas matam
os parasitas extracelulares. O tratamento é mais bem sucedido
durante a fase aguda - com um sucesso de 60%. Benznidazol e
Nifurtimox: drogas atualmente escolhidas. Requeridas diariamente
por até 2 meses ou mais. Hospitalização pode ser necessária
devido aos efeitos adversos |
|
Espécies de
Leishmania |
| Leishmania donovani |
Mediterrâneo, África, Índia, China, América do Sul |
humanos |
Velho
mundo: Flebótomos
(Phlebotomus sp)

(© Tom
Evans)
Novo mundo:
Lutzomyia chagasi |
Kala-Azar (febre Dum-Dum) Leishmaniose visceral |
|
Compostos antimoniais |
| Leishmania tropica |
Mediterrânea, África, Índia |
humanos |
Flebótomos
(Phlebotomus sp) |
Leishmaniose tegumentar (botão do oriente) |
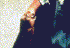
Lesão leishmânica de pele
©
Dr J.
Carl Fox,
University of Oklahoma (used with permission)
|
O único tratamento eficaz é com drogas antimoniais - efeitos
colaterais tóxicos severos. Pentostam e Glucantime
frequentemente usados. Aminosidina é eficaz em leishmaniose
cutânea e melhor tolerada. |
| Leishmania major |
África, Oriente Médio, Índia |
humanos |
Flebótomos
(Phlebotomus sp) |
Leishmaniose tegumentar
(ferida tropical) |
|
Compostos antimoniais |
| Leishmania braziliensis
L. mexicana
|
América do Sul |
humanos |
Lutzomyia sp |
Leishmaniose tegumentar- lesões de pele e de membranas mucosas,
pólipos |

Lesão de bôca
(© Tom Evans)
 Lesão de mucosa nasal (© Tom
Evans)
Lesão de mucosa nasal (© Tom
Evans)
|
Compostos antimoniais |
|
| |
|

Figura 1.
Môsca tsé-tsé: o vetor da tripanossomíase africana
©
Ohio State University, College of Biological Sciences
Figura 2
Sinal de Winterbottom
(CDC de
Parasitas na web)
|
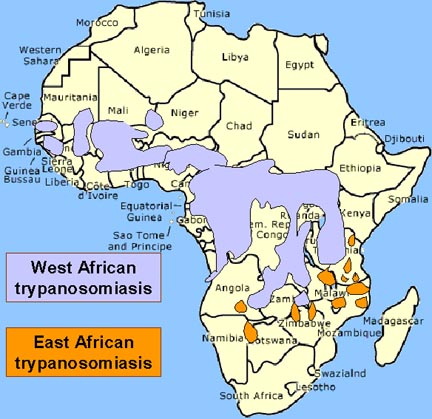
TRIPANOSSOMÍASE AFRICANA
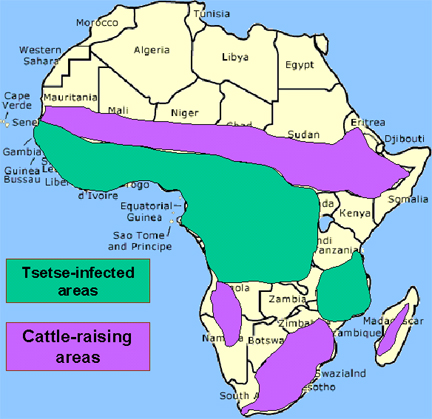
O vetor da
tripanossomíase Africana é a môsca tse-tse (figura 1) e a distribuição
dessa doença acompanha a distribuição do vetor (figura 4).
Os sintomas da
tripanossomíase Africana depende do hospedeiro e das subespécies de
tripanossoma. Nas infecções por T. gambiense ocorre estimulação
massiva do sistema immune e lise de células hospedeiras mediadas pelo
complemento (que produz anemia característica). Dor generalizada,
fraqueza, câimbras e edema dos linfonodos do pescoço (Sinal de
Winterbottom, figura 2). Parasitas invadem todos os órgãos do corpo
incluindo o coração e o SNC. Este último leva à apatia, deficiência
mental, tremores, convulsões e sono, coma. Há perda de peso rápida e
morte após alguns meses de desnutrição, parada cardíaca, pneumonia ou
uma infecção parasitária. No caso de infecções pelo T. brucei
rhodesiense, não há coma ou sintomas do sistema nervoso, visto que
os pacientes provavelmente morrem antes que esses sintomas se
desenvolvam.
Recentemente em ascenção, há um mínimo de 20.000 novos casos por ano;
50.000.000 pessoas sob risco. Nagana proíbe criação de gado em grandes
areas da África contribuindo para a desnutrição.
|
|
|
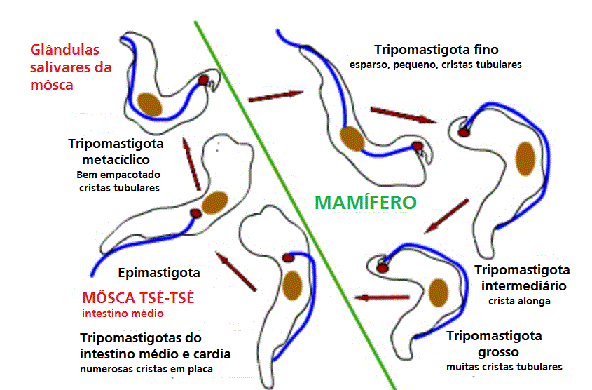
TRIPANOSSOMAS AFRICANOS
Estrutura de um Tripanossoma Africano
Tripanossomas
são protozoários unicelulares (figura 3) com um flagelo único que contém
microtúbulos em arranjo 9+2 típico de outros flagelos. Na base do
flagelo está o cinetoplasto (figura 3) que contém DNA na forma de cerca
de 6000 círculos contatenados. O DNA cinetoplástico corresponde a 10%
do DNA cellular total e é o local importante de ação de algumas drogas
anti-tripanossomas, tais como Ethidium® . O cinetoplasto é parte da
mitocôndria única e longa que muda de morfologia durante os vários
estágios do ciclo de vida.
A maioria das
outras organelas são as típicas de qualquer célula eucariótica. Na
superfície da célula existem microtúbulos peliculares sub-membranosos
que dão forma ao tripanossoma. Estes se situam suportando uma membrana
plasmática que está frequentemente coberta por uma capa na superfície
eletricamente densa (figure 3).
|
| |
|
Figura 3 |
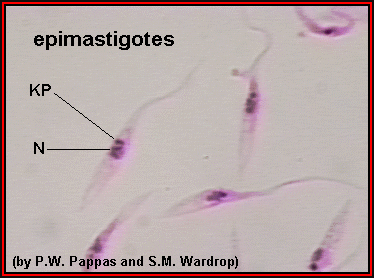
 Epimastigotas crescidos em cultura; cinetoplasto (KP) está anterior ao
núcleo (N). Na maioria das espécies de tripanossoma, este estágio
reproduz-se no intestino do vetor. ©
Universidade Estadual de Ohio, Colégio de Ciências Biológicas
Epimastigotas crescidos em cultura; cinetoplasto (KP) está anterior ao
núcleo (N). Na maioria das espécies de tripanossoma, este estágio
reproduz-se no intestino do vetor. ©
Universidade Estadual de Ohio, Colégio de Ciências Biológicas
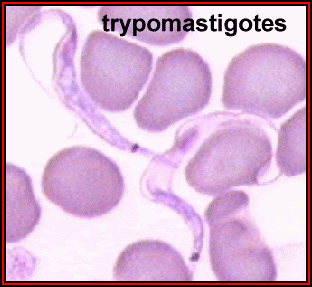
 Tripomastigotas em esfregaço sanguíneo; cinetoplasto está posterior ao
núcleo. Este estágio é encontrado em todas as espécies de tripanossomas,
e na maioria das espécies é o único estágio que reproduz em hospedeiros
vertebrados (humanos). ©
Universidade Estadual de Ohio, Colégio de Ciências Biológicas
Tripomastigotas em esfregaço sanguíneo; cinetoplasto está posterior ao
núcleo. Este estágio é encontrado em todas as espécies de tripanossomas,
e na maioria das espécies é o único estágio que reproduz em hospedeiros
vertebrados (humanos). ©
Universidade Estadual de Ohio, Colégio de Ciências Biológicas
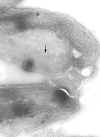
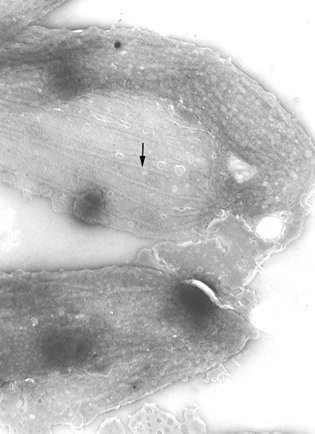
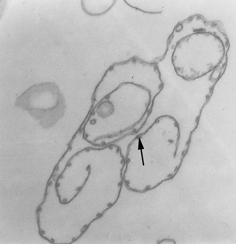
 Película membranosa de um tripanossoma. Esquerda: A membrana plasmática
foi rompida para revelar os microtúbulos (seta). Micrografia eletrônica
com coloração negativa. Direita: Micrografia eletrônica. Fina camada
mostrando microtúbulos (seta) e membrana plasmática
Película membranosa de um tripanossoma. Esquerda: A membrana plasmática
foi rompida para revelar os microtúbulos (seta). Micrografia eletrônica
com coloração negativa. Direita: Micrografia eletrônica. Fina camada
mostrando microtúbulos (seta) e membrana plasmática
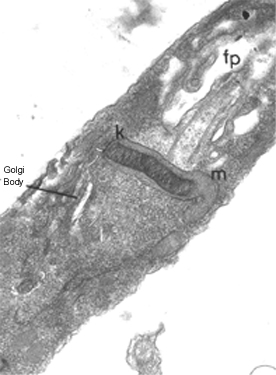
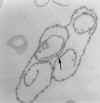
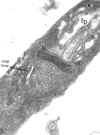 TFina
seção de um tripanossoma (Leptomonas collosoma) mostrando o
cinetoplasto (k) que é a região que contém DNA da única mitocôndria (m).
O cinetoplasto é encontrado na base do flagelo e da bolsa flagelar (fp)
TFina
seção de um tripanossoma (Leptomonas collosoma) mostrando o
cinetoplasto (k) que é a região que contém DNA da única mitocôndria (m).
O cinetoplasto é encontrado na base do flagelo e da bolsa flagelar (fp)
|
|
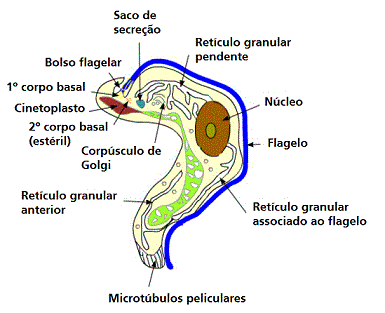
Figura 4 |
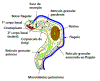 Diagrama que mostra as principais estruturas reveladas por microscopia
eletrônica da forma tripomastigota do tripanossoma salivar Trypanosoma
congolense. É mostrado corte em seções sagitais, exceto a maior parte da
fossa do flagelo e a extremidade anterior do corpo. (Adaptado de
Vickerman, K., 1969. J Protozool. 16:54-69.) (Nota: as três sub-espécies
(rodesiense, gambiense e brucei) não podem ser distinguidas
morfologicamente)
Diagrama que mostra as principais estruturas reveladas por microscopia
eletrônica da forma tripomastigota do tripanossoma salivar Trypanosoma
congolense. É mostrado corte em seções sagitais, exceto a maior parte da
fossa do flagelo e a extremidade anterior do corpo. (Adaptado de
Vickerman, K., 1969. J Protozool. 16:54-69.) (Nota: as três sub-espécies
(rodesiense, gambiense e brucei) não podem ser distinguidas
morfologicamente)
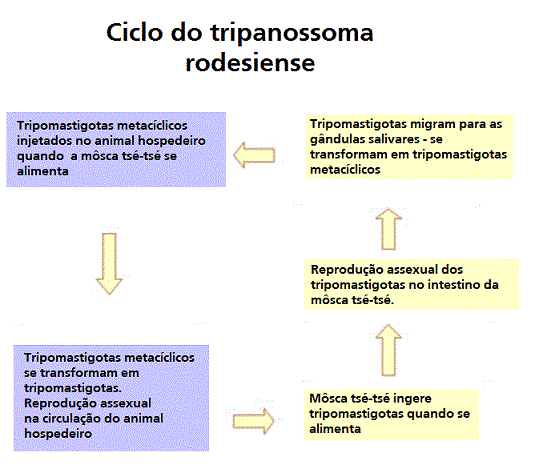
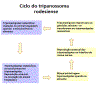 Ciclo
de vida do Tripanossoma rodesiense Ciclo
de vida do Tripanossoma rodesiense
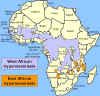 Distribuição
da Doença do Sono do Oeste da África (ou Gambiense) e do Leste da África (ou
rodesiense) Distribuição
da Doença do Sono do Oeste da África (ou Gambiense) e do Leste da África (ou
rodesiense)
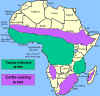 A distribuição da mosca tse-tse e áreas de criação de gado
A distribuição da mosca tse-tse e áreas de criação de gado
|
|
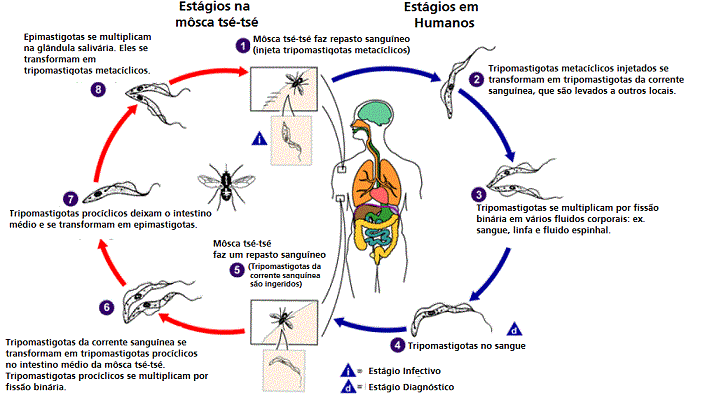
Figura
5 |
 Ciclo de vida do Trypanosoma brucei. Estágios do desenvolvimento
encontrado na corrente sanguínea do hospedeiro mamífero, o intestino
medio da tse-tse, e as glândulas salivares da tse-tse. Redesenhado de
Vickerman
Ciclo de vida do Trypanosoma brucei. Estágios do desenvolvimento
encontrado na corrente sanguínea do hospedeiro mamífero, o intestino
medio da tse-tse, e as glândulas salivares da tse-tse. Redesenhado de
Vickerman
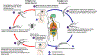 Durante um repasto sanguíneo no hospedeiro mamífero, uma môsca tse-tse
infectada (gênero Glossina) injeta tripomastigotas metacíclicos
no tecido da pele. Os parasitas entram no sistema linfático e passam
para a corrente sanguínea
Durante um repasto sanguíneo no hospedeiro mamífero, uma môsca tse-tse
infectada (gênero Glossina) injeta tripomastigotas metacíclicos
no tecido da pele. Os parasitas entram no sistema linfático e passam
para a corrente sanguínea
 .
Dentro do hospedeiro, eles se transformam em tripomastigotas da corrente
sanguínea .
Dentro do hospedeiro, eles se transformam em tripomastigotas da corrente
sanguínea
 ,
são levados a outros locais através do corpo, atingem outros fluidos
corporais (ex. linfa, fluido espinhal), e continuam a replicação por
fissão binária ,
são levados a outros locais através do corpo, atingem outros fluidos
corporais (ex. linfa, fluido espinhal), e continuam a replicação por
fissão binária
 .
O ciclo de vida inteiro dos Tripanossomas Africanos é representado por
estágios extracelulares. A môsca tse-tse se torna infectada por
tripomastigotas da corrente sanguínea quando fazem um repasto sanguíneo
em um hospedeiro mamífero infectado
( .
O ciclo de vida inteiro dos Tripanossomas Africanos é representado por
estágios extracelulares. A môsca tse-tse se torna infectada por
tripomastigotas da corrente sanguínea quando fazem um repasto sanguíneo
em um hospedeiro mamífero infectado
( , ,
 ).
No intestino médio da môsca, os parasitas se transformam em
tripomastigotas procíclicos, se multiplicam por fissão binária ).
No intestino médio da môsca, os parasitas se transformam em
tripomastigotas procíclicos, se multiplicam por fissão binária
 ,
deixam o intestimo médio e se transformam em epimastigotas ,
deixam o intestimo médio e se transformam em epimastigotas
 .
Os epimastigotas atingem as glândulas salivares da môsca e continuam se
multiplicando por fissão binária .
Os epimastigotas atingem as glândulas salivares da môsca e continuam se
multiplicando por fissão binária
 .
O ciclo na môsca dura aproximadamente 3 semanas. Humanos são o
reservatório principal do Trypanosoma brucei gambiense, mas estas
espécies podem ser também encontradas em animais. Animais selvagens são
os principais reservatórios do T. b. rhodesiense. .
O ciclo na môsca dura aproximadamente 3 semanas. Humanos são o
reservatório principal do Trypanosoma brucei gambiense, mas estas
espécies podem ser também encontradas em animais. Animais selvagens são
os principais reservatórios do T. b. rhodesiense.
DPDx, Divisão de Doenças Parasitárias CDC.
|
| |
Formas do T.
Brucei em mamíferos e insetos hospedeiros
Os vários estágios do
ciclo de vida do T. brucei em cada um de seus hospedeiros pode ser
distinguido morfologicamente (figura 5)
|
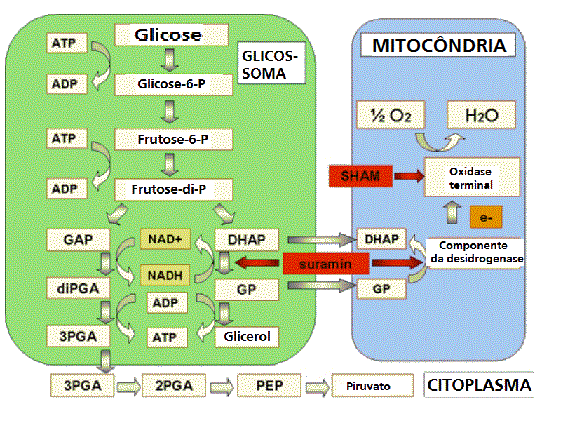
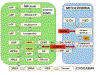 Figura
6
Via glicolítica em formas longas-delgadas do T. brucei. Os sítios
principais de inibição por drogas tripanossomicidas in vivo são
indicadas por setas vermelhas. Abreviaturas: SHAM, ácido
salicilhidroxâmico; G-6-P, glucose-6-fosfato; F-6-P, frutose-6-fosfato;
FDP, frutose-I,6-difosfato; GAP, gliceraldeído-3-fosfato; DHAP,
dihidroxicetona fosfato; GP sn-glicerol-3-fosfato; diPGA,
1,3-difosfoglicerato; 3PGA e 2PGA, 3- e 2-fosfoglicerato,
respectivamente; PEP, fosfoenolpiruvato.
Figura
6
Via glicolítica em formas longas-delgadas do T. brucei. Os sítios
principais de inibição por drogas tripanossomicidas in vivo são
indicadas por setas vermelhas. Abreviaturas: SHAM, ácido
salicilhidroxâmico; G-6-P, glucose-6-fosfato; F-6-P, frutose-6-fosfato;
FDP, frutose-I,6-difosfato; GAP, gliceraldeído-3-fosfato; DHAP,
dihidroxicetona fosfato; GP sn-glicerol-3-fosfato; diPGA,
1,3-difosfoglicerato; 3PGA e 2PGA, 3- e 2-fosfoglicerato,
respectivamente; PEP, fosfoenolpiruvato. |
Bioquímica e
biologia molecular de tripanossomas africanos
Metabolismo oxidativo
Todos os organismos
vivos fazem ATP como um carreador de energia. Este é produzido principalmente
pela oxidação de carboidratos usando a glicólise e o ciclo dos ácidos
tricarboxílicos (TCA) (figura 6). Devido ao fato de animais de vida livre (como
nós) não termos uma abundância em alimentos, nós precisamos de um ciclo TCA mais
eficiente para a maior parte de nossa produção de ATP.
O tripanossoma encontra
ambientes muito diferentes em diferentes estágios do seu ciclo de vida. Na
corrente sanguínea dos mamíferos há uma abundância de oxigênio e glicose. O
oposto acontece no intestino do inseto ou na hemolinfa. Isso se reflete no
número de reações de glicólise e ciclos do TCA que podem ocorrer na elaboração
do cinetoplasto/mitocôndria. Claramente, se um organismo pode prescindir do TCA
ele pode também prescindir da mitocôndria.
As formas de T.
brucei no intestino do inseto têm um complemento completo de enzimas do TCA
e da glicólise. Isso não é de surpreender pois nutrientes NÃO são abundantes; na
maioria dos organismos que usam fosforilação oxidativa a produção de ATP é
sensível ao cianeto porque os citocromos a/a3 reagem com cianeto e
não podem mais transferir um elétron para o oxigênio para formar água;
entretanto, a fosforilação oxidativa nas formas intestinais de insetos de
Tripanossomas é apenas PARCIALMENTE SENSÍVEL AO CIANETO. O sistema a/a3
ainda é sensível a CN- mas em Tripanossomas existe um sistema
citocromo O adicional que é insensível ao CN- . Pouco se sabe sobre o
sistema citocromo O. No inseto, a fermentação aeróbica parcial produz succinato,
piruvato, acetato e também dióxido de carbono. Prolina é uma fonte de energia
importante.
As formas de T.
brucei na corrente sanguínea de mamíferos usam somente uma glicólise
ineficiente porque há muitos nutrientes disponíveis; visto que glicólise produz
muito menos ATP do que o ciclo do TCA, a respiração é 50 vezes maior que a de
uma célula normal de mamífero , e na corrente sanguínea T. brucei usa 10
vezes mais quantidade de energia do que no intestino do inseto.
Observação das aulas de
bioquímica: Se você usa apenas a glicólise para fazer ATP como é feito na
respiração anaeróbica nos nossos músculos ou em levêdos, você não pode excretar
simplesmente piruvato, que é o que o tripanossoma faz na verdade, pois você irá
reduzir todo o seu NAD+ a NADH e a via tôda vai parar por falta de
substrato oxidado. Nós convertemos piruvato a lactato enquanto que os levêdos
convertem piruvato a etanol para oxidar nossos NADH de volta a NAD+ e
a glicólise continua. Tripanossomas excretam piruvato e têm outra forma de
converter NADH de volta a NAD+.
Em tripanossomas, o
metabolismo da di-hidroxicetona fosfato é necessário para a reoxidação do NADH.
Este é um sistema aeróbico que requer entretanto oxigênio, mas o consumo de
oxigênio é insensível a CN- e porisso não usa a cadeia do citocromo
normal. Uma desidrogenase contendo FAD ligado a oxidase contendo cobre. Este
complexo é o sistema glicerofosfato-oxidase. Tripanossomas na corrente sanguínea
dependem deste mecanismo, que mantém o NAD oxidado. O sistema é o alvo de duas
drogas tripanocidas: SURAMIN e SHAM (ácido salicílico-hidroxâmico), que é um
agente quelante. A oxidase terminal contém cobre. Essas drogas têm suas
especificidades, pois esta é uma via metabólica que os mamíferos não usam.
Há uma outro fato excêntrico no metabolismo dos carboidratos de tripanossomas.
Diferentemente da célula de mamífero, as nove primeiras reações da glicólise
estão associadas a uma organela (GLICOSSOMA). Por que tripanossomas precisam
compartimentalizar a glicólise, não está claro. A finalidade de um compartimento
é que todas as enzimas estejam concentradas em um mesmo lugar. Mas a difusão não
parece ser fator limitante em células que não têm glicossomas.
|
| |
Morfologia
do cinetoplasto
Muda com o
metabolismo
Tripomastigotas
finos na corrente sanguínea: mitocôndrias simples, poucas cristas curtas
e tubulares
Curtos e grossos
na corrente sanguínea: elaboração de mitocôndrias, síntese de enzimas
mitocondriais
Intestino médio
da môsca: elabora uma matriz de cristas em forma de placa. Mitocôndrias
se estendem anteriormente e posteriormente a partir do cinetoplasto.
Môsca
metacíclica: degeneração das mitocôndrias
|
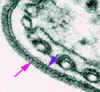 Figura 7
Eletromicrografia da capa de superfície da forma de mamífero de um
tripanossoma. A seta rosa indica a capa e a seta azul indica a membrana
plasmática
Figura 7
Eletromicrografia da capa de superfície da forma de mamífero de um
tripanossoma. A seta rosa indica a capa e a seta azul indica a membrana
plasmática
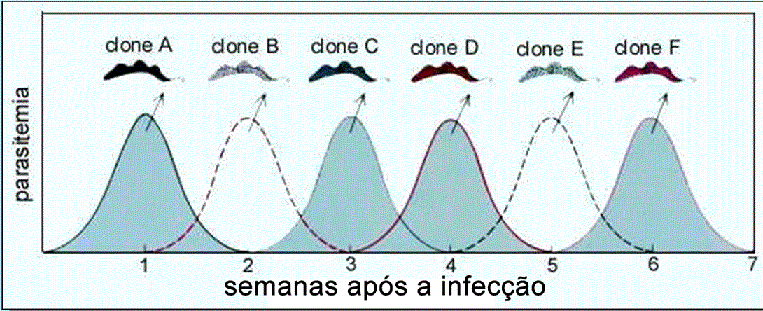
 Figura 8
Ondass sucessivas de parasitas no sangue são características da doença
do sono e se correlacionam com ciclos de febre. Uma população de
parasitas (com algumas glicoproteínas variáveis de superfície (VSGs)) se
dividem na corrente sanguínea em um període de dias. Alguns desses
tripanossomas têm VSG A na sua superfície (clone A). O sistema imune
produz anticorpos contra toda a população de antígenos e, como resultado
a maioria dos parasitas morre. Alguns tripanossomas entretanto mudam
suas capas e expressam outra VSG (ex. VSG B). Esses parasitas sobrevivem
ao expressarem um novo gene de VSG e originam uma nova população (clone
B). Na hora certa, o hospedeiro produz anticorpos contra VSG B e as
células do clone B morrem, mas novamente algumas células mudam suas
capas e sobrevivem. Este ciclo é repetido muitas vezes no curso de uma
infecção crônica pois os parasitas continuam expressando novos genes e
exibindo novos antígenos VSG. A partir de cada população sucessive é
possível se isolar tripanossomas individuais e a partir deles crescer
clones expressando determinadas VSGs.
Figura 8
Ondass sucessivas de parasitas no sangue são características da doença
do sono e se correlacionam com ciclos de febre. Uma população de
parasitas (com algumas glicoproteínas variáveis de superfície (VSGs)) se
dividem na corrente sanguínea em um període de dias. Alguns desses
tripanossomas têm VSG A na sua superfície (clone A). O sistema imune
produz anticorpos contra toda a população de antígenos e, como resultado
a maioria dos parasitas morre. Alguns tripanossomas entretanto mudam
suas capas e expressam outra VSG (ex. VSG B). Esses parasitas sobrevivem
ao expressarem um novo gene de VSG e originam uma nova população (clone
B). Na hora certa, o hospedeiro produz anticorpos contra VSG B e as
células do clone B morrem, mas novamente algumas células mudam suas
capas e sobrevivem. Este ciclo é repetido muitas vezes no curso de uma
infecção crônica pois os parasitas continuam expressando novos genes e
exibindo novos antígenos VSG. A partir de cada população sucessive é
possível se isolar tripanossomas individuais e a partir deles crescer
clones expressando determinadas VSGs.
|
Reações do
hospedeiro ao parasita
Qualquer parasita
tem problemas quando reside em outro organismo. Este último
provavelmente vai montar uma resposta imune ao parasita que poderá
destruí-lo. Alguns parasitas se escondem em superfícies que enganam o
sistema imune fazendo-se passar como células normais. Outros parasitas
(incluindo Trypansoma cruzi) entram nas célula para fugir do
sistema imune. T. brucei não faz nem um nem outro. Ao invés
disso:
O parasita é
muito antigênico, o que é uma explicação para os sintomas exibidos em um
paciente infectado. Mas o número de parasitas na corrente sanguínea não
vai aumentando, aumentando até a morte do paciente. O paciente tem ondas
de febre e ciclos na infestação pelo parasita. As ondas de parasitemia
se correlacionam com a febre observada. O número de parasitas no sangue
mostra ondas enquanto o sistema imune se recupera parcialmente da
infecção. A natureza cíclica da parasitemia é muito característica.
Então, por que
a resposta imune massiva montada pelo indivíduo infectado não elimina o
parasita como deveria? Apesar da resposta imune massiva com elevados
níveis de Ig (especialmente IgM) e profunda proliferação de linfócitos
B, deve acontecer uma mudança em alguns tripanossomas da população total
que permite que eles semeiem uma nova geração de parasitas. Mais tarde,
na fase crônica da infecção, órgãos linfóides são depletados de
linfócitos, eles engilham e zonas de fibrose substituem os linfócitos.
A imunodepressão acontece e a parasitemia se descontrola, levando à
morte.
Por que nem
todos os parasitas são destruídos por esta resposta imune tão forte?
Esta é a chave para o progesso da doença! A justificativa reside na
superfície eletronicamente densa (figura 7) que envolve as formas do
parasita na corrente sanguínea e que é o único antígeno de importância
reconhecido pelo sistema imune hospedeiro.
Se clonarmos
células isoladas de diferentes animais infectados ou pacientes, a capa é
diferente bioquimicamente! Não só um pouco diferente, mas tão diferente
que a capa de proteínas tem que ter resultado da expressão de diferentes
genes tripanossomais em cada animal. Além disso, se pegarmos células de
uma determinada onda de parasitemia do mesmo paciente, foi verificado
que todos os tripanossomas daquela onda de microrganismos estão
expressando um único antígeno de superfície, enquanto que em outras
ondas, todos os parasitas estão expressando também um único antígeno mas
sendo este antígeno completamente diferente (figura 8). Em outras
palavras, um antígeno de superfície diferente está sendo expresso. A
orla da superfície é portanto feita de ANTÍGENOS DE SUPERFÍCIE VARIÁVEIS
ou GLICOPROTEÍNAS VARIÁVEIS DE SUPERFÍCIE (VSGs). Você poderá ver o
têrmo VATs para designar tipos de antígenos variáveis.
Assim, escapar
da resposta imune depende da habilidade de expressar um novo VSG. Visto
que centenas dessas ondas de parasitemia podem ocorrer antes da morte do
paciente (em uma situação laboratorial o número de ondas é muito menor
que este) sem que nenhum antígeno seja repetido, deve haver o mesmo
número de genes VSG. Na verdade, existem provavelmente 1000-2000 desses
genes e 10% do genoma da célula é devotado a genes que expressam essas
moléculas de superfície, que permitem o organismo estar um passo avante
em relação à resposta imune do hospedeiro.
Isso levanta uma
série de perguntas: Qual a estrutura desses VSGs? Como é feita a mudança
de tipo? Como pode ser apenas um gene VSG expressado de cada vez? Como
ocorre a liberação completa do VSG?
Cada
glicoproteína de VSG tem um tamanho de cerca de 65kD com cerca de 500
aminoácidos e tem três domínios. No terminal N está a sequência sinal;
os próximos 360 aminoácidos são normalmente muito diferentes da
sequência similar em outros VSGs. Os 120 aminoácitos do terminal C são
muito semelhantes em diferentes VSGs. Esta última parte está escondida
do sistema imune por estar próxima da membrana plasmática.
A partir do sequenciamento de proteínas e do sequenciamento de cDNA nós
obtemos uma idéia diferente da porção C-terminal da molécula. O cDNA
(que dá a sequência codificada pelo gene) mostra uma sequência típica
transmembrana hidrofóbica que é usada normalmente para ligar a proteína
à membrana plasmática através de um pequeno domínio intracelular, mas o
sequenciamento da proteína mostra que a porção transmembrana da
proteína e a parte intracelular não está na proteína madura. Estas são
substituídas por uma estrutura estranha que contém açúcares,
etanolamina, fosfoinositol e ácidos graxos. Esta estrutura é comum a
todos os VSGs e é altamente antigênica quando purificada, mas não in
vivo. Isso sugere que os VSGs da capa estão empacotados para excluir
anticorpos. Portanto, este sítio não tem utilidade no desenvolvimento de
vacinas.
|
 Figura 9
Quando um tripanossoma africano expressa uma nova capa, esta está
frequentemente associada com o aparecimento de uma nova cópia do gene
daquela capa. Isso se chama de cópia ligada à expressão.
Figura 9
Quando um tripanossoma africano expressa uma nova capa, esta está
frequentemente associada com o aparecimento de uma nova cópia do gene
daquela capa. Isso se chama de cópia ligada à expressão. |
Como ocorre a
expressão sequencial das VSGs?
Digestão por endonucleases
de restrição, eletroforese em gel e Southern blot nos permite fragmentar o
genoma de tripanossomas em pedaços. Podemos então detectar um gene determinado
no genoma que apareça no nosso gel pela hibridização dele com uma sonda
complementar. Esta última normalmente é um cDNA produzido por um molde de RNAm
ou de um gene clonado.
Se formos bem sucedidos
em nossa fragmentação e se existe somente um gene no genoma para uma determinada
proteína, nós veremos apenas uma banda de hibridação no gel (contanto que uma
endonuclease não tenha clivado o gene). Tais análises mostraram que cada gene
VSG era normalmente expresso apenas uma vez no genoma, o que não é de
surpreender pois genes VSG constituem uma determinada porção do genoma e cópias
múltiplas iriam ocupar uma porção do genoma muito grande.
Nós poderemos isolar DNA
de organismos da primeira, segunda, terceira e quarta onda de parasitemia
(clones A, B, C e D). Podemos então hibridizar estes DNAs após fragmentação com
uma sonta que detecte o gene expressado na segunda onda, que está no clone B.
Encontraremos que os clones A, C e D (que não estão expressando VSG B) têm
apenas uma cópia do VSG B.
Entretando, um resultado
muito surpreendente foi encontrado, como mostrado no diagrama da esquerda
(figura 9), quando usamos uma sonda para o gene que codifica para VSG B em
células que estão expressando VSG B.
|
| |
Há uma cópia extra do gene
que está sendo expresso neste clone específico de tripanossomas. É APENAS O GENE
QUE ESTÁ SENDO EXPRESSADO QUE OCORRE EM UMA CÓPIA EXTRA! OU SEJA, TEMOS UMA
DUPLICAÇÃO GÊNICA mas ela ocorre apenas temporariamente pois quando o
tripanossoma muda para uma nova VSG a cópia extra normalmente desaparece e é
substituída por outra CÓPIA LIGADA À EXPRESSÃO (ELC). É a cópia extra que está
sendo transcrita em RNAm para ser traduzida em proteína, e não a cópia que está
permanentemente no genoma. A nova cópia está em um SÍTIO DE EXPRESSÃO. Sítios de
expressão estão sempre perto de um TELÔMERO, que está perto do término de um
cromossoma. Uma ELC e um gene permanente podem estar em cromossomas diferentes.
Isso sugere um mecanismo de cópia/translocação para um cassete de informação. Às
vezes, uma cópia ligada à expressão não é produzida e a cópia permanente é
transcrita. Esses genes não ELC transcritos estão sempre nos telômeros. A cópia
temporária de um gene e o uso da cópia é muito incomum.
Podem os genes ser
expressos em qualquer ordem? Existe uma sequência de expressão pré-programada?
Poderia se suspeitar que não, visto que existem milhares de genes VSG e a
capacidade de se usar todos eles seria uma vantagem seletiva. De fato, a ordem
não é absoluta. No início da fase infectiva, VSGs são produzidas por parasitas
nas glândulas salivares do inseto. Um subconjunto do repertório (cêrca de 12)
das VSGs são produzidas aí. Isso se mantém durante a primeira onda de
parasitemia no mamífero. O repertório completo é então aberto para expressão e
há uma ordem de expressão preferida mas não uma ordem fixa de expressão. Cada
gene pode ser expresso apenas uma vez durante uma infecção. Note que o fato de
que há um sub-conjunto inicial de VSGs que são expressos traz esperança para uma
vacina.
RNAm de tripanossomas
são também muito diferentes. Quase todos os RNAm observados começam com os
mesmos 35 nucleotídeos. Estes são codificados por um exon distante do resto do
gene.
Desfolhando a
capa de VSG
Será claramente
importante que o parasita desfolhe toda esta capa em um determinado momento para
sobreviver à resposta imune que está sendo desenvolvida contra sua VSG original.
Vimos antes que a forma da VSG na superfície da célula é ligada através de uma
sequência transmembrana glicolipídica e não protêica. Isso pode dar um
indicativo de como ocorre o desfolhamento.Uma fosfolipase C específica para o
tripanossoma e para a VSG é encontrada nas formas na corrente sanguínea. Assim,
uma vez que todas as VSGs têm a mesma estrutura de ligação, uma enzima é
necessária para a clivagem rápida e completa.
Possibilidades de quimioterapia
A existência de possivelmente 2000 genes de VSG torna uma vacina de largo
espectro muito improvável. Infelizmente, os antígenos comuns estão enterrados e
escondidos da resposta imune. Talvez um agente que interaja com a sequência de
35 nucleotídeos dos RNAm que são encontrados em todos os tripanossomas mas não
nos RNAm do animal hospedeiro seria eficiente, mas nada se sabe sobre isso. A
melhor possibilidade é uma vacina contra os mais ou menos 12 antígenos iniciais
que são expressados nos organismos que são injetados pela môsca tsé-tsé.
Alternativamente, um inibidor de fosfolipase C poderia ser um candidato muito
bom.
|
| |
Extraordinária edição de RNA pelos tripanossomas: O mistéria dos genes
desaparecidos
Esses organismos
têm sido demonstrados como bioquimicamente esquisitos sob muitos
aspectos, mas quando alguns dos achados em tripanossomas são observados
em outras espécies, estes processos aparentemente peculiares são
verificados como não tão peculiares afinal.
Os tripanossomas
têm um genoma muito estranho. Os cromossomas não condensam na divisão
nuclear e por isso não podemos saber quantos cromossomos existem. Os
genes nucleares incluem 1000-2000 genes que codificam para os antígenos
variáveis de superfície que permitem que a orla dos organismos seja
modificada regularmente para que ele possa evitar a resposta imune. A
maneira como isso é feito é extraordinário e envolve a mudança de uma
nova cópia de um gene para um sítio de expressão quando isso for
necessário. Até 10% do genoma é composto desses genes de antígenos
variáveis de superfície.
Além disso,
existe um cinetoplasto. Esta estrutura rica em DNA se posiciona na base
do flagelo e está em um dos terminais de uma única e longa mitocôndria
do flagelado. Ela é equivalente ao DNA mitocondrial de todas as outras
células, mas ela corresponde a uma proporção muito maior do DNA da
célula do que ocupa o DNA mitocondrial de círculo único de outras
células. Lembrem-se de que nossas mitocôndrias podem codificar para
algumas de suas próprias proteínas (algumas subunidades do citocromo e
do ribossoma) juntamente com todos os RNAs mitocondriais e ribossomais
e todos os RNAs transportadores mitocondriais. Embora o DNA do
cinetoplasto seja muito mais elaborado e ocupe uma proporção maior do
DNA da célula, ele não codifica para nenhum RNA ou proteína a mais do
que outros DNAs mitocondriais. De fato, alguns dos RNAt do cinetoplasto
não são codificados por este DNA e precisam ser importados do
citoplasma, o que não é o caso das mitocôndrias de mamíferos. O
mecanismo desta importação nao é conhecido.
A ração por que
o DNA do cinetoplasto faz tamanha proporção do DNA total das células é a
sua complexidade.
Ele contém 20-50
cópias de um MAXICÍRCULO de 22kb que é equivalente ao DNA mitocondrial
em qualquer outra mitocôndria. MAS, além disso, existem até 10,0000
MINICÍRCULOS de 1kb de função até recentemente desconhecida. E eles são
ainda mais estranhos: eles formam uma rêde de círculos concatenados.
Como dito antes, entre as coisas que são codificadas pelo maxicírulo de
DNA estão o RNA ribossomal da mitocôndria e uma variedade de enzimas da
cadeia respiratória da mitocôndria. Há também algumas ORFs (fases de
leitura abertas) não identificadas no maxicírculo de DNA.
|
| |
O enigma da
citocromo-oxidase de alguns tripanossomas, especialmente Trypansosma
brucei
Em todos os
tripanossomas que têm sido estudados, a subunidade III (CO III) da
citocromo-oxidase é codificada no DNA cinetoplástico. Todos, quer dizer,
exceto T. brucei não só fazem tudo mas T. brucei têm seus
genes para COIII no DNA cinetoplástico, mas eles estão todos na mesma
posição dentro daquele DNA. Já com a exceção do gene COIII, o
DNAk de T. brucei parece muito mais com o dos outros dois
tripanossomas Crithidia fasciculata e Leishmania tarentolae.
A única
verdadeira diferença é a ausência do gene para COIII. Nas outras duas
espécies, este gene está a montante do gene do apocitocromo b. Significa
isso que o T. brucei não tem a subunidade III da
citocromo-oxidase? Não, ele tem que ter pois ele tem que realizar a
fosforilação oxidativa. Além disso, o RNAm e a proteína estão claramente
no organismo. Talvez este gene simplesmente não esteja no mesmo lugar
que o gene similar em outros tripanossomas. Mas nós não o encontramos em
lugar nenhum. Sondagem de DNA do núcleo e do citoplasma mostra que não
existe o gene. Como podemos ter uma proteína e um RNAm sem ter um gene
que os codifique?
Visto que em
outros tripanossomas que têm claramente o gene para COIII ele está
sempre no mesmo lugar, talvez devêssemos observar melhor esta região do
genoma do cinetoplasto! Então o que existe nessa região do DNAk de T.
brucei? Existem algumas fases abertas de leitura que podem codificar
para pequenas proteínas mas nada do tamanho de uma proteína COIII e, em
qualquer caso, as duas maiores têm que iniciar em um códon AUG e porisso
não podem codificar para uma proteína verdadeira. E mesmo se elas
codificassem elas iriam codificar para uma proteína muito estranha que
seria muito rica em aminoácidos carregados pois o DNA nesta região é
muito rico em G-C.
Como já foi
dito, investigadores têm procurado o gene para COIII em outro lugar do
DNAk maxicírculo e não encontraram. O DNAk em maxicírculos e
minicírculos agora está sequenciado. Usando sondas que detectam o gene
de COIII em L. tarentolae e C. fasciculata, não
encontramos nenhum gene COIII aparente em maxicírulo ou DNA nuclear de
T. brucei.
Isso quer dizer
que o gene para COIII está faltando OU é altamente divergente tanto que
as sondas não pegam. Mas, novamente, como dito acima, ele não pode
faltar pois o T. brucei tem um sistema de citocromo sensível a
cianeto semelhante aos outros dois. E não seria de se esperar que ele
fosse tão divergente em virtude do extremo grau de conservação dos
genes de COIII em todos os outros tripanossomas que têm sito estudados.
Se você não pode
encontrar o gene, você pode pensar em observar os transcritos, ou seja,
os RNAm.
Foram
encontradas evidências de um transcrito para um RNAm em T. brucei
que tem uma sequência similar à do gene de COIII em outros
tripanossomas. A similaridade das sequências permite a determinação da
fase aberta de leitura correta para o transcrito de T. brucei.
Dos 181
aminoácidos preditos pela sequência, 135 são conservados entre os três.
Se pegarmos substituições conservativas e as conservadas em uma outra
espécie nós encontramos que 160 de 181 são conservadas. Este nível de
conservação (88%) é ligeiramente melhor do que entre genes em
maxicírculos de T. brucei e L. tarentolae onde a
conservação varia de 65% a 84%.
|
| |
Isso significa
que deve haver um gene para a proteína COIII em T. brucei, mas onde ele
está?E aí, a
pergunta novamente: Há uma sequência genômica que combine com o transcrito de
COIII?
Antes foi mostrado que a
hibridização heteróloga usando sondas de COIII de L. tarentolae e C.
fasciculata probes não detectou sequências de COIII em genes de T.
brucei. Isso foi confirmado por análises por Southern blot usando sondas que
foram preditas a partir da sequência que foi obtida para o transcrito. As sondas
não hibridizam com DNAk ou com DNA total.
Nenhuma detecção foi
encontrada com sondas que detectariam 0.5 cópias do gene por genoma.
Visto que não há gene
aparentemente para esses transcritos em in T. brucei, duas possibilidades
existem:
1. O transcrito foi
feito por processamento que juntou pequenos fragmentos do RNA transcrito de
regões múltiplas. Há afinal de contas precedentes de mini-exons no sistema de
tripanossomas. Há dados que tendem a excluir esta possibilidade.
2. Existe uma intensa
edição do transcrito após ou durante a transcrição, de forma que o transcrito
final não se parece nem de longe com o gene de onde foi transcrito.
Agora, vamos voltar ao
genoma do cinetoplasto. O gene TEM que estar lá, só que não conseguimos
detectá-lo.
E o lugar para procurar
pelo gene de COIII seria imediatamente a montante do gene do apo-citocromo b,
onde o gene de COIII estaria localizado em tantos outros tripanossomas.
A sequência nesta
posição que nós vimos não contém fases abertas de leitura grandes, cujos códons
de iniciação tenham sido reinvestigados. Mas, de fato, a sequência desta região
combina com a sequência do transcrito exatamente, EXCETO pela presença de
uridinas no transcrito que não estão no DNA do gene. Aviso: os códons de
iniciação são ATG e isso explica a ausência de códons de iniciação no gene. A
ausência de 25% dos nucleotídeos explicam as fases abertas de leitura menores.
Isso também explica o alto conteúdo de G-C.
Em virtude das
diferenças entre as sequências de RNA e DNA, apenas o número e posições das Us
são afetados; Cs, Gs, e As ocorrem na mesma sequência no RNAm e no DNA genômico.
Dos 626 nucleotídeos
sequenciados inicialmente nos transcritos de COIII de T. brucei COIII,
347 são URIDINAS que não são codificadas pelo gene. Elas são adicionadas em 121
locais diferentes.
Em um desses locais, a
adição de uma U cria um códon de parada, exatamente onde o códon de parada
nativo ocorre nos outros genes de COIII.
Além disso, 16 uridinas,
preditas pela sequência genomica em 7 locais aparecem como deletadas.
A porção do transcrito
codificadora da proteína contém 315 adições e 15 deleções em 546 nucleotídeos.
Assim, 58% da parte
codificadora do RNAm resulta de edição e não está no gene original.
A edição é a razão pela
qual sondas de cDNA não hibridizam com o DNA genômico.
Para que esta edição?
Uma sugestão para essa
edição é que ela pode servir como um mecanismo de controle durante o ciclo de
vida complexo do parasita. É claro, não usando Us o gene economiza espaço. Será?
Como ocorre a edição?
O que determina onde devem ser colocadas ou retiradas as Us? Existe um
transcrito original que corresponda à sequência genômica do cinetoplasto?
A primeira etapa é de
fato fazer um transcrito que seja complementar à sequência genômica. Este é
então editado para colocar ou retirar uridinas em locais específicos. A
sequência de um ácido nuclêico é normalmente determinada por outro ácido
nuclêico e uma classe de RNAs inteiramente nova que é até o presente própria de
tripanossomas, é usada para isso. Esses pequenos RNAs são chamados RNA-guias
(RNAg) e são codificados por regiões do maxicírculo e do minicírculo, cujas
funções não eram conhecidas previamente.
Um RNAg tem uma ponta 5' que é complementar a uma pequena região do transcrito
de RNAm inicial da COIII não editada (perto da ponta 3'). No final desta região
do RNAg haverá uma ou mais adenosinas, que não estão no transcrito primário,
seguidas de mais sequência e de uma cauda puli-U. No local de não
complementaridade o transcrito primário é cortado por uma endonuclease e uma U
(da cauda puli-U) é transferida (pela U-terminal transferase). Esta é agora
complementar à A no RNAg. Se houver outra A no RNAg, outra U será adicionada ao
transcrito primário e isso continua até que não haja outra A na sequência. Este
é o sinal para uma enzima ligase ligar o terminal 5' original do transcrito
primário à U (ou Us) recentemente inseridas na ponta 5' da porção 3' do
transcrito primário. Presumívelmente agora o RNAg se dissocia. Mas agora uma
nova sequência contendo Us está presente no transcrito primário inicialmente
editado e este é reconhecido por um RNAg diferente, que pode continuar editando
de 3' para 5' do transcrito original, e intermediários no processamento foram
encontrados, o que apóia esta idéia. Qualquer que seja o mecanismo nós temos uma
situação extraordinária, em que a maioria das uridinas do gene são retiradas e
adicionadas após a transcrição do RNAm, usando o potencial de codificação de
outro grupo de genes que codificam para RNAgs. Isso parece muito ineficiente e
foi encontrado apenas em tripanossomas e nesse nível.
|

Figura 10
Vetor: Triatoma infestans (insetos assassinos) e espécies relacionadas
e gêneros (ex. Rhodnius e Panstrongylus)
©
Ohio State University, College of Biological Sciences
|
TRIPANOSSOMAS AMERICANOS
Trypanosoma
cruzi
Doença de Chagas
A doença é
transmitida por insetos reduvídeos, incluindo os insetos assassinos e
rodnios (figura 10) que infectam o paciente quando defeca após
realizarem repasto sanguíneo.
Os sintomas da
doença de Chagas são: infecção crônica, desordens neurológicas
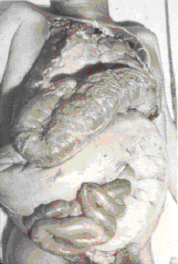
(incluindo demência), megacólon (figura 11), megaesôfago, e lesões na
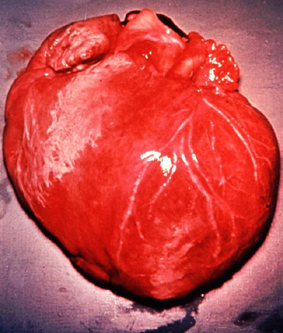
musculatura cardíaca (figura 11). A doença de Chagas é frequentemente
fatal, a menos que seja tratada.
Na doença agura ocorre sempre anemia severa, dor muscular e desordens
neurológicas. Estas últimas são comuns em crianças menores de 2 anos,
casos em que a morte acontece em cerca de um mês. A doença crônica pode
ser amena, e às vezes assintomática, mas pode haver danos nos nervos
provocando cessação das contrações musculares, batimentos cardíacos
irregulares e destruição de centros motores do sistema nervoso central.
A forma crônica da doença é encontrada em adultos mas muito
provavelmente provém de uma infecção na infância. T. cruzi pode
cruzar a barreira placentária e assim mães cronicamente infectadas podem
infectar seus bebês, que podem morrer com uma forma muito aguda da
doença.
|
|
Figura 11 |
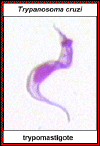 Tripomastigota de
T. cruzi
Tripomastigota de
T. cruzi
©
Ohio State University, College of Biological Sciences
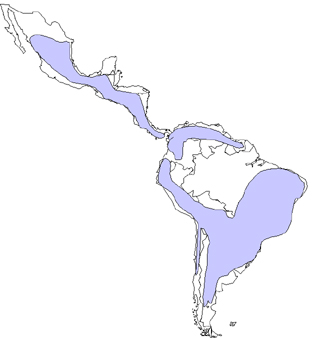
 Distribuição mundial da doença de Chagas
Distribuição mundial da doença de Chagas
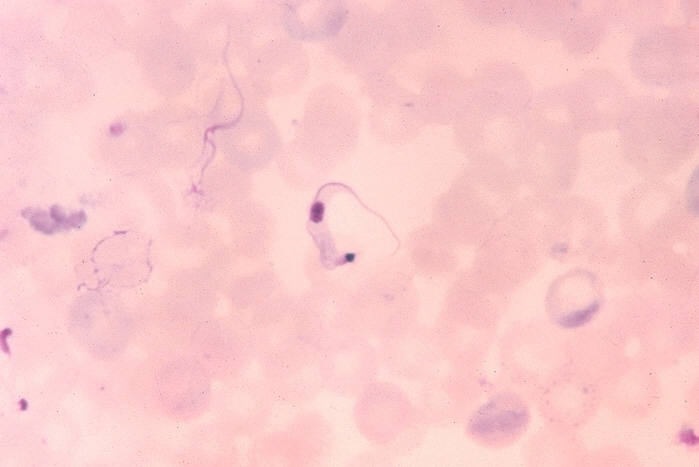
 Trypanosoma cruzi em esfregaço sanguíneo. CDC
Trypanosoma cruzi em esfregaço sanguíneo. CDC
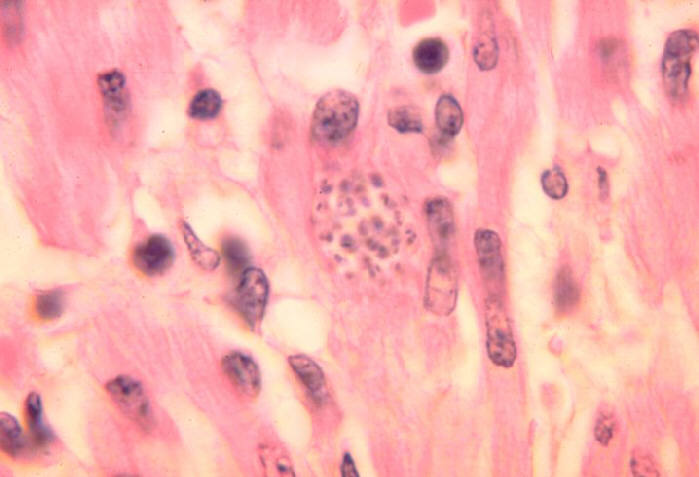
 Amastigotas (pseudocisto) de T. cruzi no coração de um cachorro.
Amastigotas (pseudocisto) de T. cruzi no coração de um cachorro.
©
Ohio
State University, College of Biological Sciences
|
| |
 Esta criança do Panama está sofrendo de doença de Chagas manifestada
como uma infecção aguda com edema no olho direito
Esta criança do Panama está sofrendo de doença de Chagas manifestada
como uma infecção aguda com edema no olho direito
 Trypanosoma cruzi no coração de um macaco.
Trypanosoma cruzi no coração de um macaco.
CDC/Dr. L.L. Moore, Jr. 1969
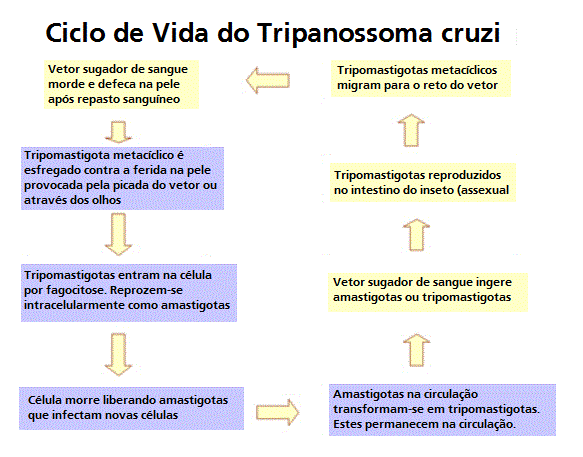
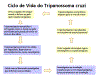 Ciclo de vida do Trypanosoma cruzi
Ciclo de vida do Trypanosoma cruzi
 MMegacólon
em paciente com doença de Chagas
MMegacólon
em paciente com doença de Chagas
 Dilatação por cardiomegalia causada pelo T. cruzi (D. Despommier
in
Parasitas na web)
Dilatação por cardiomegalia causada pelo T. cruzi (D. Despommier
in
Parasitas na web)
|
|
|
Assim como os
tripanossomas africanos, o T. cruzi escapa da resposta imune
hospedeira, mas eles não fazem isso pela mudança da capa antigênica.
Porisso, eles não têm variação antigênica. Ao invés disso, eles escapam se
escondendo no interior das células. A doença começa após uma picada do
inseto vetor. Após 7-14 dias os tripanossomas chegam nos linfonodos onde se
dividem. Aí eles formam agregados chamados pseudocistos. Quando pseudocistos
se rompem, os parasitas liberados podem entrar nas células de várias partes
do corpo, incluindo tecido linfático, músculo e tecidos ao redor dos
gânglios nervosos. A invasão do gângio nervoso cardíaco é a causa de muitas
doenças cardíacas em áreas onde o T. Cruzi é encontrado.
Ligação e
infecção da célula hospedeira
Ligação
Amastigotas se ligam
à superfície celular (ex. um monócito) via uma variedade de proteínas
receptoras incluindo fibronectina. O ácido siálico é importante pois células
deficientes em ácido siálico não são penetradas. Curiosamente, T. cruzi
tem uma enzima chamada de trans-sialidase, que na verdade coloca mais ácido
siálico nas células, consequentemente aumentando a sua captação.
Entrada na
célula
A captação é feita
por um processo de fagocitose induzida. Lisossomas migram para a superfície
quando as células entram em contato com T. cruzi e o parasita entra
em um vacúolo citoplasmático, o vacúolo parasitóforo, que é formado de um
lisossoma. Assim, agentes que induzem a migração de lisossomas de áreas
perinucleares para que eles se alinham próximos à membrana plasmática,
aumentam a infectividade. Ao contrário, o bloqueio das funções lisossomais
páram a infecção. De alguma forma, os parasitas escapam do potencial
destrutivo do lisossoma e após cerca de uma hora o T. cruzi libera
uma proteína tóxica que se insere na membrana a um pH baixo típico desta
organela. Como resultado o T. cruzi escapa para o citoplasma com a
destruição da membrana lisossomal.
|
| |
| |
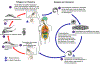 Um vetor triatomídeo (ou "bicho beijador") faz um repasto sanguíneo e
libera tripomastigotas nas suas fezes próximo do local da ferida da
picada. Tripomastigotas entram no hospedeiro através da ferida ou
através das membranas mucosas intactas, tais como a conjuntiva
.
Espécies de vetores triatomídeos da tripanossomíase pertencem aos
gêneros Triatoma, Rhodinius, e Panstrongylus.
Dentro do hospedeiro, os tripomastigotas invadem células, onde se
diferenciam em amastigotas intracelulares
.
Os amastigotas se multiplicam por fissão binária
e se diferenciam em tripomastigotas, e são então liberados na
circulação como tripomastigotas da corrente sanguínea
.
Tripomastigotas infectam células de uma variedade de tecidos e se
transformam em amastigotas intracelulares em novos locais de infecção.
As manifestações clínicas podem advir desse ciclo infectivo. Os
tripomastigotas da corrente sanguínea não se replicam (diferentemente
dos tripanossomas africanos). A replicação recomeça somente quando os
parasitas entram em outra célula ou são ingeridos por outro vetor. O
inseto se infecta ao se alimentar de sangue humano ou de animal que
contém parasitas circulantes
.
Os tripomastigotas ingeridos se transformam em epimastigotas no
intestino médio do vetor
.
Os parasitas se multiplicam e se diferenciam no intestino médio
e se diferenciam em tripomastigotas metacíclicos infectivos no intestino
terminal
.
Um vetor triatomídeo (ou "bicho beijador") faz um repasto sanguíneo e
libera tripomastigotas nas suas fezes próximo do local da ferida da
picada. Tripomastigotas entram no hospedeiro através da ferida ou
através das membranas mucosas intactas, tais como a conjuntiva
.
Espécies de vetores triatomídeos da tripanossomíase pertencem aos
gêneros Triatoma, Rhodinius, e Panstrongylus.
Dentro do hospedeiro, os tripomastigotas invadem células, onde se
diferenciam em amastigotas intracelulares
.
Os amastigotas se multiplicam por fissão binária
e se diferenciam em tripomastigotas, e são então liberados na
circulação como tripomastigotas da corrente sanguínea
.
Tripomastigotas infectam células de uma variedade de tecidos e se
transformam em amastigotas intracelulares em novos locais de infecção.
As manifestações clínicas podem advir desse ciclo infectivo. Os
tripomastigotas da corrente sanguínea não se replicam (diferentemente
dos tripanossomas africanos). A replicação recomeça somente quando os
parasitas entram em outra célula ou são ingeridos por outro vetor. O
inseto se infecta ao se alimentar de sangue humano ou de animal que
contém parasitas circulantes
.
Os tripomastigotas ingeridos se transformam em epimastigotas no
intestino médio do vetor
.
Os parasitas se multiplicam e se diferenciam no intestino médio
e se diferenciam em tripomastigotas metacíclicos infectivos no intestino
terminal
.
Trypanosoma cruzi
pode também ser transmitido através de transfusões de sangue,
transplante de órgãos, transplacentariamente, e em acidentes
laboratoriais.
DPDx, Divisão de Doenças Parasitárias CDC.
|
|
|
 Voltar à Seção de Parasitologia de Microbiologia e Imunologia On-line
Voltar à Seção de Parasitologia de Microbiologia e Imunologia On-line
Esta página foi modificada em
Monday, January 23, 2017
Página mantida por
Richard Hunt
|

 Figura
6
Via glicolítica em formas longas-delgadas do T. brucei. Os sítios
principais de inibição por drogas tripanossomicidas in vivo são
indicadas por setas vermelhas. Abreviaturas: SHAM, ácido
salicilhidroxâmico; G-6-P, glucose-6-fosfato; F-6-P, frutose-6-fosfato;
FDP, frutose-I,6-difosfato; GAP, gliceraldeído-3-fosfato; DHAP,
dihidroxicetona fosfato; GP sn-glicerol-3-fosfato; diPGA,
1,3-difosfoglicerato; 3PGA e 2PGA, 3- e 2-fosfoglicerato,
respectivamente; PEP, fosfoenolpiruvato.
Figura
6
Via glicolítica em formas longas-delgadas do T. brucei. Os sítios
principais de inibição por drogas tripanossomicidas in vivo são
indicadas por setas vermelhas. Abreviaturas: SHAM, ácido
salicilhidroxâmico; G-6-P, glucose-6-fosfato; F-6-P, frutose-6-fosfato;
FDP, frutose-I,6-difosfato; GAP, gliceraldeído-3-fosfato; DHAP,
dihidroxicetona fosfato; GP sn-glicerol-3-fosfato; diPGA,
1,3-difosfoglicerato; 3PGA e 2PGA, 3- e 2-fosfoglicerato,
respectivamente; PEP, fosfoenolpiruvato.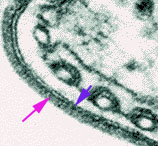 Figura 7
Eletromicrografia da capa de superfície da forma de mamífero de um
tripanossoma. A seta rosa indica a capa e a seta azul indica a membrana
plasmática
Figura 7
Eletromicrografia da capa de superfície da forma de mamífero de um
tripanossoma. A seta rosa indica a capa e a seta azul indica a membrana
plasmática
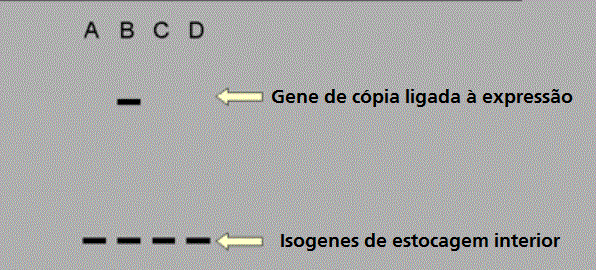 Figura 9
Quando um tripanossoma africano expressa uma nova capa, esta está
frequentemente associada com o aparecimento de uma nova cópia do gene
daquela capa. Isso se chama de cópia ligada à expressão.
Figura 9
Quando um tripanossoma africano expressa uma nova capa, esta está
frequentemente associada com o aparecimento de uma nova cópia do gene
daquela capa. Isso se chama de cópia ligada à expressão.